Medical Information


Delivery, Face Presentation, and Brow Presentation: Understanding Fetal Positions and Birth Scenarios
Introduction:.
During childbirth, the position of the baby plays a significant role in the delivery process. While the most common fetal presentation is the head-down position (vertex presentation), variations can occur, such as face presentation and brow presentation. This comprehensive article aims to provide a thorough understanding of delivery, face presentation, and brow presentation, including their definitions, causes, complications, and management approaches.
Delivery Process:
- Normal Vertex Presentation: In a typical delivery, the baby is positioned head-down, with the back of the head (occiput) leading the way through the birth canal.
- Engagement and Descent: Prior to delivery, the baby's head engages in the pelvis and gradually descends, preparing for birth.
- Cardinal Movements: The baby undergoes a series of cardinal movements, including flexion, internal rotation, extension, external rotation, and restitution, which facilitate the passage through the birth canal.
Face Presentation:
- Definition: Face presentation occurs when the baby's face is positioned to lead the way through the birth canal instead of the vertex (head).
- Causes: Face presentation can occur due to factors such as abnormal fetal positioning, multiple pregnancies, uterine abnormalities, or maternal pelvic anatomy.
- Complications: Face presentation is associated with an increased risk of prolonged labor, difficulties in delivery, increased fetal malposition, birth injuries, and the need for instrumental delivery.
- Management: The management of face presentation depends on several factors, including the progression of labor, the size of the baby, and the expertise of the healthcare provider. Options may include closely monitoring the progress of labor, attempting a vaginal delivery with careful maneuvers, or considering a cesarean section if complications arise.
Brow Presentation:
- Definition: Brow presentation occurs when the baby's head is partially extended, causing the brow (forehead) to lead the way through the birth canal.
- Causes: Brow presentation may result from abnormal fetal positioning, poor engagement of the fetal head, or other factors that prevent full flexion or extension.
- Complications: Brow presentation is associated with a higher risk of prolonged labor, difficulty in descent, increased chances of fetal head entrapment, birth injuries, and the potential need for instrumental delivery or cesarean section.
- Management: The management of brow presentation depends on various factors, such as cervical dilation, progress of labor, fetal size, and the presence of complications. Close monitoring, expert assessment, and a multidisciplinary approach may be necessary to determine the safest delivery method, which can include vaginal delivery with careful maneuvers, instrumental assistance, or cesarean section if warranted.
Delivery Techniques and Intervention:
- Obstetric Maneuvers: In certain situations, skilled healthcare providers may use obstetric maneuvers, such as manual rotation or the use of forceps or vacuum extraction, to facilitate delivery, reposition the baby, or prevent complications.
- Cesarean Section: In cases where vaginal delivery is not possible or poses risks to the mother or baby, a cesarean section may be performed to ensure a safe delivery.
Conclusion:
Delivery, face presentation, and brow presentation are important aspects of childbirth that require careful management and consideration. Understanding the definitions, causes, complications, and appropriate management approaches associated with these fetal positions can help healthcare providers ensure safe and successful deliveries. Individualized care, close monitoring, and multidisciplinary collaboration are crucial in optimizing maternal and fetal outcomes during these unique delivery scenarios.
Hashtags: #Delivery #FacePresentation #BrowPresentation #Childbirth #ObstetricDelivery
On the Article

Krish Tangella MD, MBA

Alexander Enabnit

Alexandra Warren
Please log in to post a comment.
Related Articles
Test your knowledge, asked by users, related centers, related specialties, related physicians, related procedures, related resources, join dovehubs.
and connect with fellow professionals
Related Directories
At DoveMed, our utmost priority is your well-being. We are an online medical resource dedicated to providing you with accurate and up-to-date information on a wide range of medical topics. But we're more than just an information hub - we genuinely care about your health journey. That's why we offer a variety of products tailored for both healthcare consumers and professionals, because we believe in empowering everyone involved in the care process. Our mission is to create a user-friendly healthcare technology portal that helps you make better decisions about your overall health and well-being. We understand that navigating the complexities of healthcare can be overwhelming, so we strive to be a reliable and compassionate companion on your path to wellness. As an impartial and trusted online resource, we connect healthcare seekers, physicians, and hospitals in a marketplace that promotes a higher quality, easy-to-use healthcare experience. You can trust that our content is unbiased and impartial, as it is trusted by physicians, researchers, and university professors around the globe. Importantly, we are not influenced or owned by any pharmaceutical, medical, or media companies. At DoveMed, we are a group of passionate individuals who deeply care about improving health and wellness for people everywhere. Your well-being is at the heart of everything we do.
For Patients
For professionals, for partners.
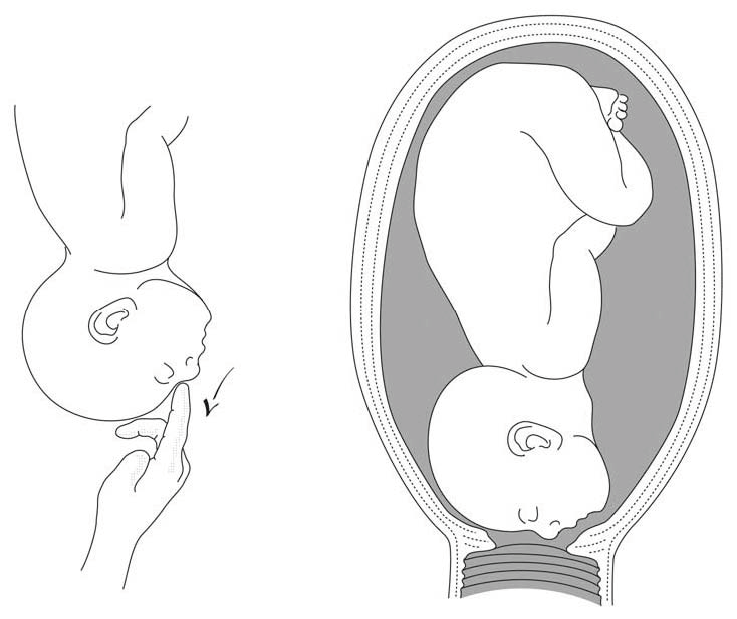
7.10 Brow presentation
Brow presentation constitutes an absolute foeto-pelvic disproportion, and vaginal delivery is impossible (except with preterm birth or extremely low birth weight).
This is an obstetric emergency, because labour is obstructed and there is a risk of uterine rupture and foetal distress.
7.10.1 Diagnosis
- Head is high; as with a face presentation, there is a cleft between the head and back, but it is less marked.
- the chin (it is not a face presentation),
- the posterior fontanelle (it is not a vertex presentation).
Figures 7.9 - Brow presentation

Any mobile presenting part can subsequently flex. The diagnosis of brow presentation is, therefore, not made until after the membranes have ruptured and the head has begun to engage in a fixed presentation. Some brow presentations will spontaneously convert to a vertex or, more rarely, a face presentation.
During delivery, the presenting part is slow to descend: the brow is becoming impacted.
7.10.2 Management
Foetus alive.
- Perform a caesarean section. When performing the caesarean section, an assistant must be ready to free the head by pushing it upward with a hand in the vagina.
- Convert the brow presentation to a face presentation: between contractions, insert the fingers through the cervix and move the head, encouraging extension (Figures 7.10).
- Attempt internal podalic version ( Section 7.9 ).
Both these manoeuvres pose a significant risk of uterine rupture. Vacuum extraction, forceps and symphysiotomy are contra-indicated.
Foetus dead
Perform an embryotomy if the cervix is sufficiently dilated (Chapter 9, Section 9.7 ) otherwise, a caesarean section.
Face and Brow Presentation
- Author: Teresa Marino, MD; Chief Editor: Carl V Smith, MD more...
- Sections Face and Brow Presentation
- Mechanism of Labor
- Labor Management
At the onset of labor, assessment of the fetal presentation with respect to the maternal birth canal is critical to the route of delivery. At term, the vast majority of fetuses present in the vertex presentation, where the fetal head is flexed so that the chin is in contact with the fetal thorax. The fetal spine typically lies along the longitudinal axis of the uterus. Nonvertex presentations (including breech, transverse lie, face, brow, and compound presentations) occur in less than 4% of fetuses at term. Malpresentation of the vertex presentation occurs if there is deflexion or extension of the fetal head leading to brow or face presentation, respectively.
In a face presentation, the fetal head and neck are hyperextended, causing the occiput to come in contact with the upper back of the fetus while lying in a longitudinal axis. The presenting portion of the fetus is the fetal face between the orbital ridges and the chin. The fetal chin (mentum) is the point designated for reference during an internal examination through the cervix. The occiput of a vertex is usually hard and has a smooth contour, while the face and brow tend to be more irregular and soft. Like the occiput, the mentum can present in any position relative to the maternal pelvis. For example, if the mentum presents in the left anterior quadrant of the maternal pelvis, it is designated as left mentum anterior (LMA).
In a brow presentation, the fetal head is midway between full flexion (vertex) and hyperextension (face) along a longitudinal axis. The presenting portion of the fetal head is between the orbital ridge and the anterior fontanel. The face and chin are not included. The frontal bones are the point of designation and can present (as with the occiput during a vertex delivery) in any position relative to the maternal pelvis. When the sagittal suture is transverse to the pelvic axis and the anterior fontanel is on the right maternal side, the fetus would be in the right frontotransverse position (RFT).
Face presentation occurs in 1 of every 600-800 live births, averaging about 0.2% of live births. Causative factors associated with a face presentation are similar to those leading to general malpresentation and those that prevent head flexion or favor extension. Possible etiology includes multiple gestations, grand multiparity, fetal malformations, prematurity, and cephalopelvic disproportion. At least one etiological factor may be identified in up to 90% of cases with face presentation.
Fetal anomalies such as hydrocephalus, anencephaly, and neck masses are common risk factors and may account for as many as 60% of cases of face presentation. For example, anencephaly is found in more than 30% of cases of face presentation. Fetal thyromegaly and neck masses also lead to extension of the fetal head.
A contracted pelvis or cephalopelvic disproportion, from either a small pelvis or a large fetus, occurs in 10-40% of cases. Multiparity or a large abdomen can cause decreased uterine tone, leading to natural extension of the fetal head.
Face presentation is diagnosed late in the first or second stage of labor by examination of a dilated cervix. On digital examination, the distinctive facial features of the nose, mouth, and chin, the malar bones, and particularly the orbital ridges can be palpated. This presentation can be confused with a breech presentation because the mouth may be confused with the anus and the malar bones or orbital ridges may be confused with the ischial tuberosities. The facial presentation has a triangular configuration of the mouth to the orbital ridges compared to the breech presentation of the anus and fetal genitalia. During Leopold maneuvers, diagnosis is very unlikely. Diagnosis can be confirmed by ultrasound evaluation, which reveals a hyperextended fetal neck. [ 1 , 2 ]
Brow presentation is the least common of all fetal presentations and the incidence varies from 1 in 500 deliveries to 1 in 1400 deliveries. Brow presentation may be encountered early in labor but is usually a transitional state and converts to a vertex presentation after the fetal neck flexes. Occasionally, further extension may occur resulting in a face presentation.
The causes of a persistent brow presentation are generally similar to those causing a face presentation and include cephalopelvic disproportion or pelvic contracture, increasing parity and prematurity. These are implicated in more than 60% of cases of persistent brow presentation. Premature rupture of membranes may precede brow presentation in as many as 27% of cases.
Diagnosis of a brow presentation can occasionally be made with abdominal palpation by Leopold maneuvers. A prominent occipital prominence is encountered along the fetal back, and the fetal chin is also palpable; however, the diagnosis of a brow presentation is usually confirmed by examination of a dilated cervix. The orbital ridge, eyes, nose, forehead, and anterior fontanelle are palpated. The mouth and chin are not palpable, thus excluding face presentation. Fetal ultrasound evaluation again notes a hyperextended neck.
As with face presentation, diagnosis is often made late in labor with half of cases occurring in the second stage of labor. The most common position is the mentum anterior, which occurs about twice as often as either transverse or posterior positions. A higher cesarean delivery rate occurs with a mentum transverse or posterior [ 3 ] position than with a mentum anterior position.
The mechanism of labor consists of the cardinal movements of engagement, descent, flexion, internal rotation, and the accessory movements of extension and external rotation. Intuitively, the cardinal movements of labor for a face presentation are not completely identical to those of a vertex presentation.
While descending into the pelvis, the natural contractile forces combined with the maternal pelvic architecture allow the fetal head to either flex or extend. In the vertex presentation, the vertex is flexed such that the chin rests on the fetal chest, allowing the suboccipitobregmatic diameter of approximately 9.5 cm to be the widest diameter through the maternal pelvis. This is the smallest of the diameters to negotiate the maternal pelvis. Following engagement in the face presentation, descent is made. The widest diameter of the fetal head negotiating the pelvis is the trachelobregmatic or submentobregmatic diameter, which is 10.2 cm (0.7 cm larger than the suboccipitobregmatic diameter). Because of this increased diameter, engagement does not occur until the face is at +2 station.
Fetuses with face presentation may initially begin labor in the brow position. Using x-ray pelvimetry in a series of 7 patients, Borrell and Ferstrom demonstrated that internal rotation occurs between the ischial spines and the ischial tuberosities, making the chin the presenting part, lower than in the vertex presentation. [ 4 , 5 ] Following internal rotation, the mentum is below the maternal symphysis, and delivery occurs by flexion of the fetal neck. As the face descends onto the perineum, the anterior fetal chin passes under the symphysis and flexion of the head occurs, making delivery possible with maternal expulsive forces.
The above mechanisms of labor in the term infant can occur only if the mentum is anterior and at term, only the mentum anterior face presentation is likely to deliver vaginally. If the mentum is posterior or transverse, the fetal neck is too short to span the length of the maternal sacrum and is already at the point of maximal extension. The head cannot deliver as it cannot extend any further through the symphysis and cesarean delivery is the safest route of delivery.
Fortunately, the mentum is anterior in over 60% of cases of face presentation, transverse in 10-12% of cases, and posterior only 20-25% of the time. Fetuses with the mentum transverse position usually rotate to the mentum anterior position, and 25-33% of fetuses with mentum posterior position rotate to a mentum anterior position. When the mentum is posterior, the neck, head and shoulders must enter the pelvis simultaneously, resulting in a diameter too large for the maternal pelvis to accommodate unless in the very preterm or small infant.
Three labor courses are possible when the fetal head engages in a brow presentation. The brow may convert to a vertex presentation, to a face presentation, or remain as a persistent brow presentation. More than 50% of brow presentations will convert to vertex or face presentation and labor courses are managed accordingly when spontaneous conversion occurs.
In the brow presentation, the occipitomental diameter, which is the largest diameter of the fetal head, is the presenting portion. Descent and internal rotation occur only with an adequate pelvis and if the face can fit under the pubic arch. While the head descends, it becomes wedged into the hollow of the sacrum. Downward pressure from uterine contractions and maternal expulsive forces may cause the mentum to extend anteriorly and low to present at the perineum as a mentum anterior face presentation.
If internal rotation does not occur, the occipitomental diameter, which measures 1.5 cm wider than the suboccipitobregmatic diameter and is thus the largest diameter of the fetal head, presents at the pelvic inlet. The head may engage but can descend only with significant molding. This molding and subsequent caput succedaneum over the forehead can become so extensive that identification of the brow by palpation is impossible late in labor. This may result in a missed diagnosis in a patient who presents later in active labor.
If the mentum is anterior and the forces of labor are directed toward the fetal occiput, flexing the head and pivoting the face under the pubic arch, there is conversion to a vertex occiput posterior position. If the occiput lies against the sacrum and the forces of labor are directed against the fetal mentum, the neck may extend further, leading to a face presentation.
The persistent brow presentation with subsequent delivery only occurs in cases of a large pelvis and/or a small infant. Women with gynecoid pelvis or multiparity may be given the option to labor; however, dysfunctional labor and cephalopelvic disproportion are more likely if this presentation persists.
Labor management of face and brow presentation requires close observation of labor progression because cephalopelvic disproportion, dysfunctional labor, and prolonged labor are much more common. As mentioned above, the trachelobregmatic or submentobregmatic diameters are larger than the suboccipitobregmatic diameter. Duration of labor with a face presentation is generally the same as duration of labor with a vertex presentation, although a prolonged labor may occur. As long as maternal or fetal compromise is not evident, labor with a face presentation may continue. [ 6 ] A persistent mentum posterior presentation is an indication for delivery by cesarean section.
Continuous electronic fetal heart rate monitoring is considered mandatory by many authors because of the increased incidence of abnormal fetal heart rate patterns and/or nonreassuring fetal heart rate patterns. [ 7 ] An internal fetal scalp electrode may be used, but very careful application of the electrode must be ensured. The mentum is the recommended site of application. Facial edema is common and can obscure the fetal facial anatomy and improper placement can lead to facial and ophthalmic injuries. Oxytocin can be used to augment labor using the same precautions as in a vertex presentation and the same criteria of assessment of uterine activity, adequacy of the pelvis, and reassuring fetal heart tracing.
Fetuses with face presentation can be delivered vaginally with overall success rates of 60-70%, while more than 20% of fetuses with face presentation require cesarean delivery. Cesarean delivery is performed for the usual obstetrical indications, including arrest of labor and nonreassuring fetal heart rate pattern.
Attempts to manually convert the face to vertex (Thom maneuver) or to rotate a posterior position to a more favorable anterior mentum position are rarely successful and are associated with high fetal morbidity and mortality and maternal morbidity, including cord prolapse, uterine rupture, and fetal cervical spine injury with neurological impairment. Given the availability and safety of cesarean delivery, internal rotation maneuvers are no longer justified unless cesarean section cannot be readily performed.
Internal podalic version and breech extraction are also no longer recommended in the modern management of the face presentation. [ 8 ]
Operative delivery with forceps must be approached with caution. Since engagement occurs when the face is at +2 position, forceps should only be applied to the face that has caused the perineum to bulge. Increased complications to both mother and fetus can occur [ 9 ] and operative delivery must be approached with caution or reserved when cesarean section is not readily available. Forceps may be used if the mentum is anterior. Although the landmarks are different, the application of any forceps is made as if the fetus were presenting directly in the occiput anterior position. The mouth substitutes for the posterior fontanelle, and the mentum substitutes for the occiput. Traction should be downward to maintain extension until the mentum passes under the symphysis, and then gradually elevated to allow the head to deliver by flexion. During delivery, hyperextension of the fetal head should be avoided.
As previously mentioned, the persistent brow presentation has a poor prognosis for vaginal delivery unless the fetus is small, premature, or the maternal pelvis is large. Expectant management is reasonable if labor is progressing well and the fetal well-being is assessed, as there can be spontaneous conversion to face or vertex presentation. The earlier in labor that brow presentation is diagnosed, the higher the likelihood of conversion. Minimal intervention during labor is recommended and some feel the use of oxytocin in the brow presentation is contraindicated.
The use of operative vaginal delivery or manual conversion of a brow to a more favorable presentation is contraindicated as the risks of perinatal morbidity and mortality are unacceptably high. Prolonged, dysfunctional, and arrest of labor are common, necessitating cesarean section delivery.
The incidence of perinatal morbidity and mortality and maternal morbidity has decreased due to the increased incidence of cesarean section delivery for malpresentation, including face and brow presentation.
Neonates delivered in the face presentation exhibit significant facial and skull edema, which usually resolves within 24-48 hours. Trauma during labor may cause tracheal and laryngeal edema immediately after delivery, which can result in neonatal respiratory distress. In addition, fetal anomalies or tumors, such as fetal goiters that may have contributed to fetal malpresentation, may make intubation difficult. Physicians with expertise in neonatal resuscitation should be present at delivery in the event that intubation is required. When a fetal anomaly has been previously diagnosed by ultrasonographic evaluation, the appropriate pediatric specialists should be consulted and informed at time of labor.
Bellussi F, Ghi T, Youssef A, et al. The use of intrapartum ultrasound to diagnose malpositions and cephalic malpresentations. Am J Obstet Gynecol . 2017 Dec. 217 (6):633-41. [QxMD MEDLINE Link] .
[Guideline] Ghi T, Eggebø T, Lees C, et al. ISUOG Practice Guidelines: intrapartum ultrasound. Ultrasound Obstet Gynecol . 2018 Jul. 52 (1):128-39. [QxMD MEDLINE Link] . [Full Text] .
Shaffer BL, Cheng YW, Vargas JE, Laros RK Jr, Caughey AB. Face presentation: predictors and delivery route. Am J Obstet Gynecol . 2006 May. 194(5):e10-2. [QxMD MEDLINE Link] .
Borell U, Fernstrom I. The mechanism of labour. Radiol Clin North Am . 1967 Apr. 5(1):73-85. [QxMD MEDLINE Link] .
Borell U, Fernstrom I. The mechanism of labour in face and brow presentation: a radiographic study. Acta Obstet Gynecol Scand . 1960. 39:626-44.
Gardberg M, Leonova Y, Laakkonen E. Malpresentations--impact on mode of delivery. Acta Obstet Gynecol Scand . 2011 May. 90(5):540-2. [QxMD MEDLINE Link] .
Collaris RJ, Oei SG. External cephalic version: a safe procedure? A systematic review of version-related risks. Acta Obstet Gynecol Scand . 2004 Jun. 83(6):511-8. [QxMD MEDLINE Link] .
Verspyck E, Bisson V, Gromez A, Resch B, Diguet A, Marpeau L. Prophylactic attempt at manual rotation in brow presentation at full dilatation. Acta Obstet Gynecol Scand . 2012 Nov. 91(11):1342-5. [QxMD MEDLINE Link] .
Johnson JH, Figueroa R, Garry D. Immediate maternal and neonatal effects of forceps and vacuum-assisted deliveries. Obstet Gynecol . 2004 Mar. 103(3):513-8. [QxMD MEDLINE Link] .
Benedetti TJ, Lowensohn RI, Truscott AM. Face presentation at term. Obstet Gynecol . 1980 Feb. 55(2):199-202. [QxMD MEDLINE Link] .
BROWNE AD, CARNEY D. OBSTETRICS IN GENERAL PRACTICE. MANAGEMENT OF MALPRESENTATIONS IN OBSTETRICS. Br Med J . 1964 May 16. 1(5393):1295-8. [QxMD MEDLINE Link] .
Campbell JM. Face presentation. Aust N Z J Obstet Gynaecol . 1965 Nov. 5(4):231-4. [QxMD MEDLINE Link] .

Contributor Information and Disclosures
Teresa Marino, MD Assistant Professor, Attending Physician, Division of Maternal-Fetal Medicine, Tufts Medical Center Disclosure: Nothing to disclose.
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference Disclosure: Received salary from Medscape for employment. for: Medscape.
Carl V Smith, MD The Distinguished Chris J and Marie A Olson Chair of Obstetrics and Gynecology, Professor, Department of Obstetrics and Gynecology, Senior Associate Dean for Clinical Affairs, University of Nebraska Medical Center Carl V Smith, MD is a member of the following medical societies: American College of Obstetricians and Gynecologists , American Institute of Ultrasound in Medicine , Association of Professors of Gynecology and Obstetrics , Central Association of Obstetricians and Gynecologists , Society for Maternal-Fetal Medicine , Council of University Chairs of Obstetrics and Gynecology , Nebraska Medical Association Disclosure: Nothing to disclose.
Chitra M Iyer, MD, Perinatologist, Obstetrix Medical Group, Fort Worth, Texas.
Chitra M Iyer, MD is a member of the following medical societies: American College of Obstetricians and Gynecologists , Society of Maternal-Fetal Medicine .
Disclosure: Nothing to disclose.
What would you like to print?
- Print this section
- Print the entire contents of
- Print the entire contents of article
- HIV in Pregnancy
- Pulmonary Disease and Pregnancy
- Kidney Disease and Pregnancy
- Vaccinations/Immunizations During Pregnancy
- Anemia and Thrombocytopenia in Pregnancy
- Common Pregnancy Complaints and Questions
- Adrenal Disease and Pregnancy
- Is immunotherapy for cancer safe in pregnancy?
- Labetalol, Nifedipine: Outcome on Pregnancy Hypertension
- Olympic Moms Are Redefining Exercise in Pregnancy

- Drug Interaction Checker
- Pill Identifier
- Calculators

- 2020/viewarticle/immunotherapy-cancer-safe-pregnancy-2024a100083dnews news Is immunotherapy for cancer safe in pregnancy?

- 2002261369-overviewDiseases & Conditions Diseases & Conditions Postterm Pregnancy
- 2001/viewarticle/labetalol-nifedipine-outcome-pregnancy-hypertension-2024a1000capnews news Labetalol, Nifedipine: Outcome on Pregnancy Hypertension

- 2022 New Pearls of Exxcellence Articles
Management of Brow, Face, and Compound Malpresentations
Author: Meera Kesavan, MD
Mentor: Lisa Keder MD Editor: Daniel JS Martingano DO MBA PhD
Registered users can also download a PDF or listen to a podcast of this Pearl. Log in now , or create a free account to access bonus Pearls features.
Fetal malpresentation, including brow, face, or compound presentations, complicates around 3-4% of all term births. Because these abnormal fetal presentations still are cephalic, many such cases result in vaginal deliveries, yet there are increased risks for adverse outcomes, including cesarean delivery resultant surgical complications, persistent malpresentation precluding vaginal delivery, and abnormal labor resulting in arrest of dilation or descent.
These fetal malpresentation are differentiated in the following ways:
- In face presentations, the presenting part is the mentum, which is further divided based on its position, including mentum posterior, mentum transverse or mentum anterior positions. This typically occurs because of hyperextension of the neck and the occiput touching the fetal back. Mentum anterior malpresentations can potentially achieve vaginal deliveries, whereas mentum posterior malpresentations cannot.
- In brow presentations, there is less extension of the fetal neck as in face presentations making the leading fetal part being the area between the anterior fontanelle and the orbital ridges. These presentations are uncommon and are managed similarly to face presentations. Brow presentation can be further described based on the position of the anterior fontanelle as frontal anterior, posterior, or transverse.
- Compound presentation is defined as the leading fetal part, including a fetal extremity, alongside a cephalic or breech presentation. Management of compound presentations is expected (and often incidentally noted following delivery) because the extremity will often either retract as the head descends or will feasibly allow for delivery in its current position, with manipulation attempts to reduce the compound presentation usually avoided.
Risk factors for brow and face presentations include fetal CNS malformations, congenital or chromosomal anomalies, advanced maternal age, low birthweight, abnormal maternal pelvic anatomy (e.g. contracted pelvis, cephalopelvic disporotion, platypelloid pelvis, etc.) and nulliparity. non-Hispanic White women have the highest risk for malpresentation, whereas non-Hispanic Black women have the lowest risk.
Diagnosis usually is made during the second stage of labor while performing routine vaingla examinations and involves palpation of the abnormal leading fetal part (forehead, orbital ridge, orbits, nose, etc.) Obstetric ultrasound can additionally provide complimentary information to support these diagnoses and distinguish from other fetal malpresentations or malpositions. In face presentation, the mentum (chin) and mouth are palpable.
Management considerations for face, brow, and compounds presentations are unique with compound presentations having higher rates of vaginal delivery and lower complications as compared to either brow or face presentations.
- For brow presentations, approximately 30-40% of brow presentations will convert to a face presentation, and about 20% will convert to a vertex presentation. Anterior positions have the possibility of vaginal deliveries and can be managed by usual labor management principles, whereas mentum posterior positions are indications for cesarean delivery.
- For face presentations, the likelihood of vaginal delivery depends on the orientation of the mentum, with mentum anterior being most suitable for vaginal delivery. If the fetus is mentum posterior, flexion of the neck is precluded and results in the inability of fetal descent.
- For compound presentations, management is expectant and manipulation of the leading extremities should be avoided. Most cases of compound presentation result in vaginal deliveries. For term deliveries, compound presentations with parts other than the hand are unlikely to result in safe vaginal delivery.
Labor management for brow and face presentation overall involves continuous fetal heart rate monitoring and repeat clinical assessments, given the increased potential of fetal complications as noted. Caution should be used with internal monitoring devices, which can cause ophthalmic injury or trauma to the presenting fetal parts, with the use of fetal scalp electrodes discouraged and intrauterine pressure catheters acceptable with appropriate clinical judgment and feasibility.
Midforceps, breech extraction, and manual manipulation are not recommended and increase the risk of maternal and neonatal morbidity.
Neonatal outcomes for both face and brow presentations include facial edema, bruising, and soft tissue trauma. Complications of compound presentation specifically include umbilical cord prolapse and injury to the presenting limb. With appropriate management, neonatal and maternal morbidity for face, brow, and compound presentations are low.
Further Reading:
Bar-El L, Eliner Y, Grunebaum A, Lenchner E, et al. Race and ethnicity are among the predisposing factors for fetal malpresentation at term. Am J Obstet Gynecol MFM. 2021 Sep;3(5):100405. doi: 10.1016/j.ajogmf.2021.100405. Epub 2021 Jun 4. PMID: 34091061.
Bellussi F, Ghi T, Youssef A, et al. The use of intrapartum ultrasound to diagnose malpositions and cephalic malpresentations. Am J Obstet Gynecol. 2017 Dec;217(6):633-641. doi: 10.1016/j.ajog.2017.07.025. Epub 2017 Jul 22. PMID: 28743440 .
Pilliod RA, Caughey AB. Fetal Malpresentation and Malposition: Diagnosis and Management. Obstet Gynecol Clin North Am. 2017 Dec;44(4):631-643. doi: 10.1016/j.ogc.2017.08.003. PMID: 29078945 .
Zayed F, Amarin Z, Obeidat B, et al. Face and brow presentation in northern Jordan, over a decade of experience. Arch Gynecol Obstet. 2008 Nov;278(5):427-30. doi: 10.1007/s00404-008-0600-0. Epub 2008 Feb 19. PMID: 18283473 .
Initial Approval: August 2013; Revised: 11/2016; Revised July 2018; Reaffirmed January 2020; Revised September 2021. Revised July 2023.
This site uses cookies.

This feature is only available for registered users.
Log in here:, not a registered user.
Becoming a registered user gives you access to special features like PDF downloads and podcast episodes of each SASGOG Pearl of Exxcellence.
Create a Free Account
Are you sure you want to remove this Pearl from your favorites list?
- Getting pregnant
- Preschooler
- Life as a parent
- Baby essentials
- Find your birth club
- Free antenatal classes
- Meet local parents & parents-to-be
- See all in Community
- Ovulation calculator
- Am I pregnant quiz
- How to get pregnant fast
- Best sex positions
- Signs of pregnancy
- How many days after your period can you get pregnant?
- How age affects fertility
- Very early signs of pregnancy
- What fertile cervical mucus looks like
- Think you're pregnant but the test is negative?
- Faint line on pregnancy test
- See all in Getting pregnant
- Pregnancy week by week
- How big is my baby?
- Due date calculator
- Baby movements week by week
- Symptoms you should never ignore
- Hospital bag checklist
- Signs of labour
- Your baby's position in the womb
- Baby gender predictor
- Vaginal spotting
- Fetal development chart
- See all in Pregnancy
- Baby names finder
- Baby name inspiration
- Popular baby names 2022
- Numerology calculator
- Gender-neutral names
- Old-fashioned names
- See all in Baby names
- Your baby week by week
- Baby milestones by month
- Baby rash types
- Baby poop chart
- Ways to soothe a crying baby
- Safe co-sleeping
- Teething signs
- Growth spurts
- See all in Baby
- Your toddler month by month
- Toddler development milestones
- Dealing with tantrums
- Toddler meals
- Food & fussy eating
- When to start potty training
- Moving from a cot to a bed
- Help your child sleep through
- Games & activities
- Vomiting: what's normal?
- See all in Toddler
- Your child month by month
- Food ideas & nutrition
- How kids learn to share
- Coping with aggression
- Bedtime battles
- Anxiety in children
- Dealing with public tantrums
- Great play ideas
- Is your child ready for school?Top tips for starting school
- See all in Preschooler
- Postnatal symptoms to watch out for
- Stitches after birth
- Postpartum blood clots
- Baby showers
- Sex secrets for parents
- See all in Life as a parent
- Best baby products
- Best formula and bottles for a windy baby
- Best car seats if you need three to fit
- Best nappies
- Best Moses baskets
- Best baby registries
- Best baby sleeping bags
- Best baby humidifier
- Best baby monitors
- Best baby bath seat
- Best baby food
- See all in Baby essentials
- Back pain in pregnancy
- Pelvic girdle pain
- Perineal massage
- Signs you're having a boy
- Signs you're having a girl
- Can you take fish oil while pregnant?
- 18 weeks pregnant bump
- Can you eat salami when pregnant?
- Edwards' syndrome
- Missed miscarriage
- Should I harvest my colostrum?
- Rhesus positive vs. Rhesus negative
- What do contractions feel like?
- Hunger in early pregnancy
- First poop after birth
- When do babies sit up?
- When can babies have salt?
- MMR vaccine rash
- Vaping while breastfeeding
- How to transition from formula to milk
- When do babies start grabbing things?
- Sperm allergy: can sperm cause itching?
- How long after taking folic acid can I get pregnant?
What is brow presentation?

- the size or shape of your pelvis
- because your baby is premature
- an abnormality that prevents your baby from tucking in her chin
- having too much amniotic fluid ( polyhydramnios )
Was this article helpful?
Forceps and ventouse (assisted birth)

Parents' tips: what to do if your mum wants to be at your baby's birth

How can I use breathing exercise during labour? (Video)

Dads' guide to pregnancy: one month

Where to go next


An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager
Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed, delivery, face and brow presentation, affiliations.
- 1 Vilnius University, Lithuania, Imperial London Healthcare NHS Trust
- 2 University of Health Sciences, Rawalpindi Medical College
- PMID: 33620804
- Bookshelf ID: NBK567727
The term presentation describes the leading part of the fetus or the anatomical structure closest to the maternal pelvic inlet during labor. The presentation can roughly be divided into the following classifications: cephalic, breech, shoulder, and compound. Cephalic presentation is the most common and can be further subclassified as vertex, sinciput, brow, face, and chin. The most common presentation in term labor is the vertex, where the fetal neck is flexed to the chin, minimizing the head circumference.
Face presentation – an abnormal form of cephalic presentation where the presenting part is mentum. This typically occurs because of hyperextension of the neck and the occiput touching the fetal back. Incidence of face presentation is rare, accounting for approximately 1 in 600 of all presentations.
In brow presentation, the neck is not extended as much as in face presentation, and the leading part is the area between the anterior fontanelle and the orbital ridges. Brow presentation is considered the rarest of all malpresentation with a prevalence of 1 in 500 to 1 in 4000 deliveries.
Both face and brow presentations occur due to extension of the fetal neck instead of flexion; therefore, conditions that would lead to hyperextension or prevent flexion of the fetal neck can all contribute to face or brow presentation. These risk factors may be related to either the mother or the fetus. Maternal risk factors are preterm delivery, contracted maternal pelvis, platypelloid pelvis, multiparity, previous cesarean section, black race. Fetal risk factors include anencephaly, multiple loops of cord around the neck, masses of the neck, macrosomia, polyhydramnios.
These malpresentations are usually diagnosed during the second stage of labor when performing a digital examination. It is possible to palpate orbital ridges, nose, malar eminences, mentum, mouth, gums, and chin in face presentation. Based on the position of the chin, face presentation can be further divided into mentum anterior, posterior, or transverse. In brow presentation, anterior fontanelle and face can be palpated except for the mouth and the chin. Brow presentation can then be further described based on the position of the anterior fontanelle as frontal anterior, posterior, or transverse.
Diagnosing the exact presentation can be challenging, and face presentation may be misdiagnosed as frank breech. To avoid any confusion, a bedside ultrasound scan can be performed. The ultrasound imaging can show a reduced angle between the occiput and the spine or, the chin is separated from the chest. However, ultrasound does not provide much predicting value in the outcome of the labor.
Copyright © 2024, StatPearls Publishing LLC.
PubMed Disclaimer
Conflict of interest statement
Disclosure: Julija Makajeva declares no relevant financial relationships with ineligible companies.
Disclosure: Mohsina Ashraf declares no relevant financial relationships with ineligible companies.
- Continuing Education Activity
- Introduction
- Anatomy and Physiology
- Indications
- Contraindications
- Preparation
- Technique or Treatment
- Complications
- Clinical Significance
- Enhancing Healthcare Team Outcomes
- Review Questions
Similar articles
- Sonographic diagnosis of fetal head deflexion and the risk of cesarean delivery. Bellussi F, Livi A, Cataneo I, Salsi G, Lenzi J, Pilu G. Bellussi F, et al. Am J Obstet Gynecol MFM. 2020 Nov;2(4):100217. doi: 10.1016/j.ajogmf.2020.100217. Epub 2020 Aug 18. Am J Obstet Gynecol MFM. 2020. PMID: 33345926
- Sonographic evaluation of the fetal head position and attitude during labor. Ghi T, Dall'Asta A. Ghi T, et al. Am J Obstet Gynecol. 2024 Mar;230(3S):S890-S900. doi: 10.1016/j.ajog.2022.06.003. Epub 2023 May 19. Am J Obstet Gynecol. 2024. PMID: 37278991 Review.
- Leopold Maneuvers. Superville SS, Siccardi MA. Superville SS, et al. 2023 Feb 19. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. 2023 Feb 19. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. PMID: 32809649 Free Books & Documents.
- Intrapartum sonographic assessment of the fetal head flexion in protracted active phase of labor and association with labor outcome: a multicenter, prospective study. Dall'Asta A, Rizzo G, Masturzo B, Di Pasquo E, Schera GBL, Morganelli G, Ramirez Zegarra R, Maqina P, Mappa I, Parpinel G, Attini R, Roletti E, Menato G, Frusca T, Ghi T. Dall'Asta A, et al. Am J Obstet Gynecol. 2021 Aug;225(2):171.e1-171.e12. doi: 10.1016/j.ajog.2021.02.035. Epub 2021 Mar 4. Am J Obstet Gynecol. 2021. PMID: 33675795
- Labor with abnormal presentation and position. Stitely ML, Gherman RB. Stitely ML, et al. Obstet Gynecol Clin North Am. 2005 Jun;32(2):165-79. doi: 10.1016/j.ogc.2004.12.005. Obstet Gynecol Clin North Am. 2005. PMID: 15899353 Review.
- Gardberg M, Leonova Y, Laakkonen E. Malpresentations--impact on mode of delivery. Acta Obstet Gynecol Scand. 2011 May;90(5):540-2. - PubMed
- Tapisiz OL, Aytan H, Altinbas SK, Arman F, Tuncay G, Besli M, Mollamahmutoglu L, Danışman N. Face presentation at term: a forgotten issue. J Obstet Gynaecol Res. 2014 Jun;40(6):1573-7. - PubMed
- Zayed F, Amarin Z, Obeidat B, Obeidat N, Alchalabi H, Lataifeh I. Face and brow presentation in northern Jordan, over a decade of experience. Arch Gynecol Obstet. 2008 Nov;278(5):427-30. - PubMed
- Bashiri A, Burstein E, Bar-David J, Levy A, Mazor M. Face and brow presentation: independent risk factors. J Matern Fetal Neonatal Med. 2008 Jun;21(6):357-60. - PubMed
- Shaffer BL, Cheng YW, Vargas JE, Laros RK, Caughey AB. Face presentation: predictors and delivery route. Am J Obstet Gynecol. 2006 May;194(5):e10-2. - PubMed
Publication types
- Search in PubMed
- Search in MeSH
- Add to Search
Related information
Linkout - more resources, full text sources.
- NCBI Bookshelf

- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.
Brow Presentation
- First Online: 02 August 2023
Cite this chapter

- Syeda Batool Mazhar 2 &
- Zahra Ahmed Muslim 2
584 Accesses
Brow presentation is the rarest of all malpresentations. Anencephaly, neck masses in fetus, polyhydramnios, multiple loops of cord around neck are the fetal factors leading to brow presentation. Contracted pelvis, preterm labour, platypelloid pelvis are some of the contributory maternal factors for brow presentation. Diagnosis is usually made during second stage of labour during prevaginal examination when anterior frontanelle and face are palpated. Cesarean section is performed in brow presentation as it is unusual to get conversion in average sized fetus once membranes have ruptured.
This is a preview of subscription content, log in via an institution to check access.
Access this chapter
Subscribe and save.
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Available as EPUB and PDF
- Compact, lightweight edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
- Durable hardcover edition
Tax calculation will be finalised at checkout
Purchases are for personal use only
Institutional subscriptions
Arulkumaran S, Robson M, editors. Munro Kerr operative obstetrics. 13th ed. Elsevier, Amsterdam; 2019. p. 89–93.
Google Scholar
Malvasi A, Barbera A, Di Vagno G, Gimovsky A, Berghella V, Ghi T, Di Renzo GC, Tinelli A. Asynclitism: a literature review of an often forgotten clinical condition. J Matern Fetal Neonatal Med. 2015;28(16):1890–4. https://doi.org/10.3109/14767058.2014.972925 . Epub 2014 Oct 29.PMID: 25283847.
Article PubMed Google Scholar
Bellussi F, Ghi T, Youssef A, Salsi G, Giorgetta F, Parma D, Simonazzi G, Gianluigi P. The use of intrapartum ultrasound to diagnose malpositions and cephalic malpresentations. Am J Obstet Gynecol. 2017;217(6):633–41.
Lanni SM, Gherman R, Gonik B. Malpresentations. Amsterdam: Elsevier; 2017.
Book Google Scholar
Bashiri A, Burstein E, Bar-David J, et al. Face and brow presentation: independent risk factors. J Matern Fetal Neonatal Med. 2008;21(6):357–60.
Hawkins JL, Koffel BL. Chapter 35. In: Chestnut’s obstetric anesthesia: principles and practice: abnormal presentation & multiple gestation. 6th ed; 2020. p. 830.
Meltzer RM, Sactleben MR, Friedman EA. Brow presentation. Obstet Gynecol Surv. 1968;23(6):255–63.
Article Google Scholar
Borell U, Fernstrom I. The mechanism of labour in face and brow presentation: a radiological study. Acta Obstet Gynecol Scand. 1960;39:626–44.
Article CAS PubMed Google Scholar
Levy DL. Persistent brow presentation: a new approach to management. South Med J. 1976;69(2):191–2.
Download references
Author information
Authors and affiliations.
MCH Centre, PIMS, Islamabad, Pakistan
Syeda Batool Mazhar & Zahra Ahmed Muslim
You can also search for this author in PubMed Google Scholar
Editor information
Editors and affiliations.
Department of Obstetrics and Gynecology, Sarojini Naidu Medical College, Agra, Uttar Pradesh, India
Ruchika Garg
Rights and permissions
Reprints and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter
Mazhar, S.B., Muslim, Z.A. (2023). Brow Presentation. In: Garg, R. (eds) Labour and Delivery. Springer, Singapore. https://doi.org/10.1007/978-981-19-6145-8_8
Download citation
DOI : https://doi.org/10.1007/978-981-19-6145-8_8
Published : 02 August 2023
Publisher Name : Springer, Singapore
Print ISBN : 978-981-19-6144-1
Online ISBN : 978-981-19-6145-8
eBook Packages : Medicine Medicine (R0)
Share this chapter
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Publish with us
Policies and ethics
- Find a journal
- Track your research
What Is Brow Presentation? What Are Its Complications?
What Is Brow Presentation?
What leads to brow presentation, diagnosis of brow presentation, how to avoid c-section if baby is in brow presentation, what complications can arise due to brow presentation.
Unlike the flexed position, in a brow presentation, the baby’s head will not be well flexed into its chest. Therefore, her head and neck will be extended back a little, as if it is looking up. If the baby remains in a brow presentation, it is doubtful that there will be enough space for the baby to descend through the pelvis. This increases the chances of a C-section . Brow presentation is least common of all fetal presentations. In fact, it happens one in every 1400 deliveries. Over half of the babies who are in brow presentation in the early labor will flex their head down during the pushing stage of the labor and the labor may progress as expected. Out of the other 50%, some babies tend to tip their head further back to the face first position while they descends further into the birth canal. Compared to the brow presentation, face first position has a higher chance to undergo a vaginal birth, provided, the chin of the baby is near the pubic bone. But if the baby’s chin is near the tailbone, C-section is the only option to avoid any complications in the delivery. In spite of the fact that brow presentation very rarely happens, it can happen to anybody. If the baby stays in a brow presentation, it is highly unlikely that there will be enough room for it to pass through the pelvis. If the labor is not progressing, or that the baby is becoming distressed, then the doctor will recommend a caesarean delivery.
There are several conditions, which increase the chances of brow presentation. The brow presentation usually takes place because of :
- Polyhydramnios : Excess amniotic fluid can make it difficult for the baby’s head to take a flexed position
- Size and shape of the pelvis: Abnormally shaped and sized pelvis can make it difficult for the baby to pick up a vertex presentation. Android pelvis, which has a triangular or heart-shaped inlet with a narrower front part, is usually behind most of the brow presentations. Similarly, contracted pelvis, a pelvis that is abnormally small, can cause brow presentation
- Fetal abnormality: Fetal abnormalities such as hydrocephalus, anencephaly and neck masses accounts for the majority of brow presentations
- Premature birth/low birth weight baby: If the baby is born prematurely or if the baby is having low birth weight , the chances of brow presentation increases
- Big baby : If the baby is larger than normal size, the baby tends to extend its head instead of curling inward
- Multiple pregnancies: Multiple pregnancies also increase the risk of brow presentation
- Multiple nuchal cords: If the umbilical cord wraps around the baby’s neck, obviously, it cannot tuck its chin into the chest. In such cases, the baby tends to be brow or face presentations
- Laxity of the uterus: If the uterine wall loses its firmness, the baby may not able to hold its chin tucked to the chest firmly and the baby tends to be in brow presentation
- Cephalopelvic disproportion (CPD): If the mother’s pelvis and the baby’s head are not proportionate to each other, brow presentation can happen

When the baby is in brow presentation, the labor will not progress as it should and prolonged labor can result in fetal distress, calling for an immediate C-section. However, if the baby picks up brow presentation and your cervix is fully dilated, there are two procedures through which the doctors try to avoid the need of C-section.
- Manual rotation: Doctor inserts his hand through the cervix and tries to flex the baby’s head
- The baby’s head should be engaged in the pelvis and should be in a front anterior position
- The pelvis should have sufficient room to permit the ventouse cup to be inserted posteriorly and to reach the occiput
- Ability and experience of the obstetrician
- How favorable is the position of the baby’s head inside the pelvis
- Available space inside the pelvis
If both these methods fail, then the doctor will go ahead with the decision to perform a caesarean.
There are several complications associated with a brow presentation if vaginal delivery is attempted without proper measures.
- Increased chances of spinal cord injury are associated with brow presentation
- Fetal distress
- Abnormal shape of the baby’s head after delivery
- Prolonged labor
- Increased chances of using forceps which in turn increases the chances of facial trauma
- Obstructed labor
If it is your first delivery, it is very unlikely that your baby will be in a brow presentation. Also if you had a brow presentation in one delivery, it doesn’t mean that it will definitely happen in your next delivery. Once you are closer to your delivery date, make sure you do not miss any of your doctor appointments.It is advisable to follow your doctor’s instructions from the very beginning of your pregnancy. Make sure you take all precautionary measures to avoid any kind of uneasiness. Have a balanced diet and sufficient rest. Keep yourself positive as you get ready for a healthy delivery . Have a safe and happy pregnancy!

With a rich experience in pregnancy and parenting, our team of experts create insightful, well-curated, and easy-to-read content for our to-be-parents and parents at all stages of parenting.
Related Posts
Meaning of advait – origin, popularity and more, meaning of sathvik – origin, popularity and more, tea during pregnancy – safe choices and risks.
Save my name, email, and website in this browser for the next time I comment.
Type above and press Enter to search. Press Esc to cancel.
- GP practice services
- Health advice
- Health research
- Medical professionals
Health topics
Advice and clinical information on a wide variety of healthcare topics.
All health topics
Latest features
Allergies, blood & immune system
Bones, joints and muscles
Brain and nerves
Chest and lungs
Children's health
Cosmetic surgery
Digestive health
Ear, nose and throat
General health & lifestyle
Heart health and blood vessels
Kidney & urinary tract
Men's health
Mental health
Oral and dental care
Senior health
Sexual health
Signs and symptoms
Skin, nail and hair health
Travel and vaccinations
Treatment and medication
Women's health
Healthy living
Expert insight and opinion on nutrition, physical and mental health.
Exercise and physical activity
Healthy eating
Healthy relationships
Managing harmful habits
Mental wellbeing
Relaxation and sleep
Managing conditions
From ACE inhibitors for high blood pressure, to steroids for eczema, find out what options are available, how they work and the possible side effects.
Featured conditions
ADHD in children
Crohn's disease
Endometriosis
Fibromyalgia
Gastroenteritis
Irritable bowel syndrome
Polycystic ovary syndrome
Scarlet fever
Tonsillitis
Vaginal thrush
Health conditions A-Z
Medicine information
Information and fact sheets for patients and professionals. Find out side effects, medicine names, dosages and uses.
All medicines A-Z
Allergy medicines
Analgesics and pain medication
Anti-inflammatory medicines
Breathing treatment and respiratory care
Cancer treatment and drugs
Contraceptive medicines
Diabetes medicines
ENT and mouth care
Eye care medicine
Gastrointestinal treatment
Genitourinary medicine
Heart disease treatment and prevention
Hormonal imbalance treatment
Hormone deficiency treatment
Immunosuppressive drugs
Infection treatment medicine
Kidney conditions treatments
Muscle, bone and joint pain treatment
Nausea medicine and vomiting treatment
Nervous system drugs
Reproductive health
Skin conditions treatments
Substance abuse treatment
Vaccines and immunisation
Vitamin and mineral supplements
Tests & investigations
Information and guidance about tests and an easy, fast and accurate symptom checker.
About tests & investigations
Symptom checker
Blood tests
BMI calculator
Pregnancy due date calculator
General signs and symptoms
Patient health questionnaire
Generalised anxiety disorder assessment
Medical professional hub
Information and tools written by clinicians for medical professionals, and training resources provided by FourteenFish.
Content for medical professionals
FourteenFish training
- Professional articles
Evidence-based professional reference pages authored by our clinical team for the use of medical professionals.
View all professional articles A-Z
Actinic keratosis
Bronchiolitis
Molluscum contagiosum
Obesity in adults
Osmolality, osmolarity, and fluid homeostasis
Recurrent abdominal pain in children
Medical tools and resources
Clinical tools for medical professional use.
All medical tools and resources
Malpresentations and malpositions
Peer reviewed by Dr Laurence Knott Last updated by Dr Colin Tidy, MRCGP Last updated 22 Jun 2021
Meets Patient’s editorial guidelines
- Download Download Article PDF has been downloaded
- Share via email
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find one of our health articles more useful.
In this article :
Malpresentation, malposition.
Usually the fetal head engages in the occipito-anterior position (more often left occipito-anterior (LOA) rather than right) and then undergoes a short rotation to be directly occipito-anterior in the mid-cavity. Malpositions are abnormal positions of the vertex of the fetal head relative to the maternal pelvis. Malpresentations are all presentations of the fetus other than vertex.
Obstetrics - the pelvis and head

Continue reading below
Predisposing factors to malpresentation include:
Prematurity.
Multiple pregnancy.
Abnormalities of the uterus - eg, fibroids.
Partial septate uterus.
Abnormal fetus.
Placenta praevia.
Primiparity.
Breech presentation
See the separate Breech Presentations article for more detailed discussion.
Breech presentation is the most common malpresentation, with the majority discovered before labour. Breech presentation is much more common in premature labour.
Approximately one third are diagnosed during labour when the fetus can be directly palpated through the cervix.
After 37 weeks, external cephalic version can be attempted whereby an attempt is made to turn the baby manually by manipulating the pregnant mother's abdomen. This reduces the risk of non-cephalic delivery 1 .
Maternal postural techniques have also been tried but there is insufficient evidence to support these 2 .
Many women who have a breech presentation can deliver vaginally. Factors which make this less likely to be successful include 3 :
Hyperextended neck on ultrasound.
High estimated fetal weight (more than 3.8 kg).
Low estimated weight (less than tenth centile).
Footling presentation.
Evidence of antenatal fetal compromise.
Transverse lie 4
When the fetus is positioned with the head on one side of the pelvis and the buttocks in the other (transverse lie), vaginal delivery is impossible.
This requires caesarean section unless it converts or is converted late in pregnancy. The surgeon may be able to rotate the fetus through the wall of the uterus once the abdominal wall has been opened. Otherwise, a transverse uterine incision is needed to gain access to a fetal pole.
Internal podalic version is no longer attempted.
Transverse lie is associated with a risk of cord prolapse of up to 20%.
Occipito-posterior position
This is the most common malposition where the head initially engages normally but then the occiput rotates posteriorly rather than anteriorly. 5.2% of deliveries are persistent occipito-posterior 5 .
The occipito-posterior position results from a poorly flexed vertex. The anterior fontanelle (four radiating sutures) is felt anteriorly. The posterior fontanelle (three radiating sutures) may also be palpable posteriorly.
It may occur because of a flat sacrum, poorly flexed head or weak uterine contractions which may not push the head down into the pelvis with sufficient strength to produce correct rotation.
As occipito-posterior-position pregnancies often result in a long labour, close maternal and fetal monitoring are required. An epidural is often recommended and it is essential that adequate fluids be given to the mother.
The mother may get the urge to push before full dilatation but this must be discouraged. If the head comes into a face-to-pubis position then vaginal delivery is possible as long as there is a reasonable pelvic size. Otherwise, forceps or caesarean section may be required.
Occipito-transverse position
The head initially engages correctly but fails to rotate and remains in a transverse position.
Alternatives for delivery include manual rotation of fetal head using Kielland's forceps, or delivery using vacuum extraction. This is inappropriate if there is any fetal acidosis because of the risk of cerebral haemorrhage.
Therefore, there must be provision for a failure of forceps delivery to be changed immediately to a caesarean. The trial of forceps is therefore often performed in theatre. Some centres prefer to manage by caesarean section without trial of forceps.
Face presentations
Face presents for delivery if there is complete extension of the fetal head.
Face presentation occurs in 1 in 1,000 deliveries 5 .
With adequate pelvic size, and rotation of the head to the mento-anterior position, vaginal delivery should be achieved after a long labour.
Backwards rotation of the head to a mento-posterior position requires a caesarean section.
Brow positions
The fetal head stays between full extension and full flexion so that the biggest diameter (the mento-vertex) presents.
Brow presentation occurs in 0.14% of deliveries 5 .
Brow presentation is usually only diagnosed once labour is well established.
The anterior fontanelle and super orbital ridges are palpable on vaginal examination.
Unless the head flexes, a vaginal delivery is not possible, and a caesarean section is required.
Further reading and references
- Hofmeyr GJ, Kulier R, West HM ; External cephalic version for breech presentation at term. Cochrane Database Syst Rev. 2015 Apr 1;(4):CD000083. doi: 10.1002/14651858.CD000083.pub3.
- Hofmeyr GJ, Kulier R ; Cephalic version by postural management for breech presentation. Cochrane Database Syst Rev. 2012 Oct 17;10:CD000051. doi: 10.1002/14651858.CD000051.pub2.
- Management of Breech Presentation ; Royal College of Obstetricians and Gynaecologists (Mar 2017)
- Szaboova R, Sankaran S, Harding K, et al ; PLD.23 Management of transverse and unstable lie at term. Arch Dis Child Fetal Neonatal Ed. 2014 Jun;99 Suppl 1:A112-3. doi: 10.1136/archdischild-2014-306576.324.
- Gardberg M, Leonova Y, Laakkonen E ; Malpresentations - impact on mode of delivery. Acta Obstet Gynecol Scand. 2011 May;90(5):540-2. doi: 10.1111/j.1600-0412.2011.01105.x.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Next review due: 21 Jun 2026
22 jun 2021 | latest version.
Last updated by
Peer reviewed by

Feeling unwell?
Assess your symptoms online for free
- Vishal's account
- Complications
Brow Presentation – An Overview

What Is Brow Presentation?
How can you get to know if your baby is in this position, what are the causes of brow presentation, how is the diagnosis made, complications of brow presentation delivery, alternatives for labor during brow presentation, precautions to take before and after labour, how will brow presentation affect your baby during labor.
Pregnancy is a beautiful experience that is also fraught with a host of complications and risks. One of them concerns the normal orientation of your baby inside your uterus, which is essential for a smooth delivery. This article will explain all about abnormal forehead presentation and its associated causes, complications, diagnosis, treatment and precautions.
Babies assume a fixed position in the uterus, that is with their chins tucked firmly into their chests. This position is ideal to exit the uterus smoothly. However, in some cases, the baby’s head and neck will extend backwards away from their chest. This is known as a brow presentation or forehead presentation. It is an extremely rare condition, occurring once in 1500 births. Brow presentation might obstruct vaginal births from occurring as there is less space for the baby to drop down towards the pelvic girdle. However, if brow presentation occurs early in labour, there is still time for them to flex their neck back to the right position. If not, labour might be hindered, causing stress for both, the mother and the baby. In these instances, your doctor might recommend a caesarean section. A brow baby tends to occur in women pregnant for the second or third time, or due to physical defects like an abnormally developed spine.
Brow babies are rarely detected before labor begins, but around half of them will shift to a face-first or crown-first presentation suitable for delivery. A brow presentation delivery will take much longer than normal, which is usually when the condition is discovered.
There are several potential reasons for your baby to assume this orientation. Some of them are:
- Fetal Size: Babies born preterm, or with low birth weights, raise the likelihood of them presenting brow first. This is also observed in large babies, who usually flex their head outwards rather than in towards their chest. Brow presentation can also be caused if your pelvic girdle and your baby’s head are disproportionate to each other.
- Polyhydramnios: Polyhydramnios is the condition in which there is too much amniotic fluid in your uterus. Thus, it might be tricky for your baby to fix their heads in the correct position.
- Multiple Pregnancy: Carrying twins or more in your womb decreases the amount of space available, making your babies take alternative positions to fit properly.
- Maternal Defects: If your pelvis is not the right shape and size, it might be difficult for your fetus to assume normal presentations. The most common cause of brow presentation is the triangle-shaped android pelvis and the atypically small contracted pelvis. Another maternal defect is a lax uterus, which is not firm enough to hold the baby in place, resulting in different presentations.
- Fetal Defects: If your baby has conditions such as anencephaly and hydrocephalus, their abnormally large heads will not be able to take the right position.
To diagnose brow presentation, an experienced doctor will be able to help. Ultrasound scans are compulsory for monitoring the situation. Your doctor might even conduct a digital examination to check the orientation of the baby’s facial features. If they find that the baby’s head does not rotate enough for a natural birth, they might recommend a caesarean section.
Several risks come with brow presentation birth. Some of them are:
- Labor time might be extended as the baby would have a hard time getting past the pelvis.
- Forceps might be required, which could cause cranial damage.
- Baby’s head shape might be altered due to difficulty while moving through the birth canal.
- Baby may go through stress during delivery as it would be difficult birth and may require a caesarean.
- Injuries may occur to the baby’s spinal cord due to trauma.
- Increased risk of cerebral hemorrhage in the baby as the head may take in damage.
As explained already, a baby in brow presentation might not have enough space to move downwards towards the cervix. If this happens, there are a few methods your doctor might implement to reduce the complications of natural birth. These methods require medical skill and enough space within the cervix to be attempted.
- Ventouse Birth: In this case, your doctor will use a small vacuum extraction device known as a ventouse to pull the baby’s head towards their chest. This method can be used even after you have begun to push.
- Manual Rotation: After the cervix undergoes complete dilation, your doctor might attempt to move the baby’s head into the correct position using their hands.
As there are several complications linked with brow presentations, here are some precautions for you to take before and after labour to have a successful pregnancy.
- Choose a doctor who is accomplished in obstetrics and gynaecology, so they are experienced in dealing with any potential outcome.
- Visit your doctor regularly, especially at the end of your third trimester.
- If you have been diagnosed with brow presentation, do not hesitate to go for a caesarean if strongly recommended by your doctor, as it dramatically reduces the risks involved.
Babies might end up with abnormally shaped heads if they go through vaginal birth with a brow presentation. However, as their heads are malleable, they will return to a normal shape in a few days. Extended labor might cause stress in your baby who has been stuck in an uncomfortable position the whole time. This might also lead to vertebral problems, so consult a paediatric osteopath if you are concerned.
Brow presentation can happen to anyone, so not encountering it in your first pregnancy does not mean you will not see during later pregnancies. Consume a balanced, nutritious diet, stay hydrated and get enough sleep. Avoiding tension and anxiety will help you stay strong for when your baby arrives.
Also Read : Preparing for Labour & Delivery – Smart Ways to Prepare for Childbirth

- RELATED ARTICLES
- MORE FROM AUTHOR

Anteverted Uterus & Pregnancy - Is It Normal?

Overcome Your Miscarriage Fears

How to Cope With Perineal Tears During Delivery

Baby With THREE Biological Parents Shocks The World!

When Can You Get an Abortion
 During Pregnancy")
Placental Abruption (Abruptio Placentae) During Pregnancy
Popular on parenting.

245 Rare Boy & Girl Names with Meanings

Top 22 Short Moral Stories For Kids

170 Boy & Girl Names That Mean 'Gift from God'

800+ Unique & Cute Nicknames for Boys & Girls
Latest posts.

90 Hilarious Winter Jokes for Kids to Make Holiday More Fun

60 Best Back to School Jokes for Kids to Start the Year With a Smile

100+ Heartfelt Thank You Messages and Quotes for Parents

150+ Step Daughter Quotes to Show Your Love
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Pan Afr Med J
Management of face presentation, face and lip edema in a primary healthcare facility case report, Mbengwi, Cameroon
Nzozone henry fomukong.
1 Microhealth Global Medical Centre, Mbengwi, Cameroon
2 Department of Medicine and Surgery, Faculty of Health Sciences University of Buea, Buea, Cameroon
Ngouagna Edwin
Mandeng ma linwa edgar, ngwayu claude nkfusai.
3 Department of Microbiology and Parasitology, Faculty of Science, University of Buea, Buea, Cameroon
4 Cameroon Baptist Convention Health Services (CBCHS), Yaoundé, Cameroon
Yunga Patience Ijang
5 Department of Public Health, School of Health Sciences, Catholic University of Central Africa, Box 1110, Yaoundé, Cameroon
Joyce Shirinde
6 School of Health Systems and Public Health, Faculty of Health Sciences, University of Pretoria Private Bag X323, Gezina, Pretoria, 0001, Pretoria, South Africa
Samuel Nambile Cumber
7 Institute of Medicine, Department of Public Health and Community Medicine (EPSO), University of Gothenburg, Box 414, SE - 405 30 Gothenburg, Sweden
8 Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
Face presentation is a rare obstetric event and most practitioners will go through their carriers without ever meeting one. Face presentation can be delivered vaginally only if the foetus is in the mentum anterior position. More than half of the cases of face presentation are delivered by caesarean section. Newborn infants with face presentation usually have severe facial oedema, facial bruising or ecchymosis. These syndromic facial features usually resolved within 24-48 hours.
Introduction
Face presentation is a rare unanticipated obstetric event characterized by a longitudinal lie and full extension of the foetal head on the neck with the occiput against the upper back [ 1 - 3 ]. Face presentation occurs in 0.1-0.2% of deliveries [ 3 - 5 ] but is more common in black women and in multiparous women [ 5 ]. Studies have shown that 60 per cent of face presentations have one or more of the following risk factors: small fetus, large fetus, high parity, previous caesarean section (CS), contracted pelvis, fetopelvic disproportion, cord around the neck multiple pregnancy, hypertensive disorders of pregnancy, polyhydramnios, uterine or nuchal cord anomaly. But 40 per cent of face presentations occur with none of these factors [ 6 , 7 ]. A vaginal birth at term is possible only if the fetus is in the mentum anterior position. More than half of cases of face presentation are delivered by caesarean section [ 4 ]. Newborn infants with face presentation usually have severe facial edema, facial bruising or ecchymosis [ 8 ]. Repeated vaginal examination to assess the presenting part and the progress of labor may lead to bruises in the face as well as damage to the eyes.
Patient and observation
Case presentation: a 21 year old primigravida at 40 weeks gestation from the last normal menstrual period referred to our facility for prolonged second stage of labor after visiting two health centres. She labored for a total of 14hrs, membrane ruptured spontaneously 12hrs before referral. Amniotic fluid was documented by midwife to be clear. She attended antenatal clinics in Mbengwi health centre 5 times, was diagnosed of hepatitis B during antenatal consultations, received no treatment. She did not do any ultrasound due to financial constraints. On examination, she was healthy, in painful distress, blood pressure 131/76mmhg, pulse 85 beats/min, temperature 37.2 o C SPO2 98%. On abdominal exams, uterus was gravid, fundal height 35cm, lie longitudinal, fetal heart rate 137bpm, cephalic presentation, engaged 2/5, with moderate contractions of 2 in 10 minutes. On vaginal examination, cervix was fully dilated, membranes ruptured, presenting part was face, mentum anterior. The conclusion made was mento-anterior face presentation ( Figure 1 ). Paturient was counseled, labor was augmented with 1 amp of oxytocin in 500ml of glucose 5% and started at 10drops/mins. Ten minutes later she delivered a male baby with Apgar score 6/10, 8/10 on the first and fifth minute. The baby weighed 3.2kg, length was 50cm, and head circumference was 41cm. Syndromic facial appearance with marked edema at the baby's lips, face and scalp was evident and he had bruising on the right nasolabial groove and right cheeks ( Figure 2 ). Physical examination of the infant's respiratory system, cardiovascular system, and his abdominal examination were normal, as was his neurological examination. Placenta was delivered by controlled cord traction 5mins later with all cotyledons. Delivery was complicated by a second degree perineal tear. Perineal tear was repaired with absorbable suture under local anaesthesia. Estimated blood lost was 350ml. baby received Hepatitis B immunoglobulins, hepatitis B vaccine and vitamin K were administered to the baby. 24 hours later, facial swellings resolved ( Figure 3 ), baby breast feeds well. Baby and mother were discharged on day 3 postpartum all fine.

Men-tum anterior face presentation

Bruising, marked lip and facial edema

Baby 24 hours later with all syndromic facial features resolved
Ethics : informed consent: written informed consent was obtained from the patient's parents for the publication of this case report.
Face presentation is a rare obstetric event and most practitioner will go through their carriers without ever meeting one [ 3 ]. We presented a case of face presentation noticed in the delivery room on digital examination in a patient with no risk factors. In a poor resource setting as ours where ultrasound is not readily available, this event is often scary and confusing to most midwives and nurses. This may prompt repeated vaginal exams for confirmation of presentation. This intend will lead to bruising of the baby's face and delay effective management [ 8 ]. Exact knowledge about the fetal position and level is important for providing the correct management of this malpresentation. When face presentation is diagnosed, around 60% of cases are in the mentum anterior position, 25% are mentum posterior and 15% are mentum transverse [ 5 ]. The patient presented the most common form of face presentation (mentum anterior). Labor was augmented, vaginal delivery was attempted and successfully conducted. Facial bruising, lip and face edema are very common complication of face presentation. These complications usually resolve within 24-48 hours [ 9 , 10 ] in this case facial edema completely resolved within 24hours ( Figure 3 ) and baby breastfeed well.
Repeated vaginal exams should be avoided when presenting part is unsure. Vaginal delivery should be attemped only on mentum anterior face presentation, in other cases, emergency ceserian section should be performed. Syndromic facial features in babies born from face presentation resolve completely within 24-48 hours.
Competing interests
The authors declare no competing interests.
Acknowledgements
We thank the participant of this study.
Authors’ contributions
NHF, NE, MMLE, NCN, YPI, FB, JS and SNC conceived the case series, assisted with the study design and participant enrollment, designed the study protocol and collected the data. NE, MMLE, NCN and SNC assisted in interpretation of results and wrote the manuscript. All authors read and approved the final manuscript.
- Face Presentation

Face it. We have a lot to learn about fetal positioning. The old paradigm is fetal positions are random. The new paradigm is that babies match the space available.
Face and brow presentations occur when baby’s spine extended until the head is shifted back so baby’s face comes through the pelvis first.
Baby may settle in a face or brow presentation before labor or they may become a face or brow presentation, usually when a posterior baby has it’s chin pushed further up by the pelvic floor during descent.
A baby who is in a face-first or forehead-first position often started as an extended (chin up) occiput posterior or occiput transverse position. Coming down on to the pelvic floor with the forehead leading then “converted” this baby’s head to the face first position.
The baby’s face may be bruised for a couple days after the birth. The brow presentation may cause a redness but only occasionally will cause a bruise.
Mobility of the pelvis and the freedom of maternal movements often help bring the face-first baby down through the pelvis with good strong, uterine surges.
But not always. Sometimes the labor can’t move baby down. Cesareans are more common, but a portion of the higher surgical rate is because time is not given to the mother to begin or continue labor, or to be out of bed for this labor. Monitoring becomes important. Expect a bit of an unusual heart rate to contraction pattern seen in these labors.
- When is Breech an Issue?
- Belly Mapping® Breech
- Flip a Breech
- When Baby Flips Head Down
- Breech & Bicornuate Uterus
- Breech for Providers
- What if My Breech Baby Doesn't Turn?
- Belly Mapping ®️ Method
- After Baby Turns
- Head Down is Not Enough
- Sideways/Transverse
- Asynclitism
- Oblique Lie
- Left Occiput Transverse
- Right Occiput Anterior
- Right Occiput Posterior
- Right Occiput Transverse
- Left Occiput Anterior
- OP Truths & Myths
- Anterior Placenta
- Body Balancing
What makes labor easier for a face-first baby and you?
Balance the body and the baby will thank you by curling into position to aim, not their face, but the crown of their head.
Flexion is physiological. So support physiology and the baby will change their position. We may need a little physics.
In Labor with a Face or Brow Presentation
Back baby up!
Forward-leaning Inversion with a jiggle of the buttocks right through 1-2 contractions often backs baby up so they can tuck their chin. Then they can aim into the pelvis with an easier position.

A little effort can make labor a lot easier!
Only after baby’s crown is first, then do Side-lying Release in labor.
Before Labor with a face or brow presentation
Face presentation may reflect a psoas/pelvic floor imbalance with a collapse in the front body.
Free the piriformis, strengthen the buttocks, lengthen the hamstrings, squat for lengthening the pelvic floor, don’t worry about strengthening the pelvic floor right now. Alignment, walking, stabilizing and lengthening will tone the pelvic floor. Use it by breathing with your whole body.
Before labor, it’s safe to do Side-lying Release when baby’s face-first head isn’t in the pelvis yet.
Free the way
The psoas is the upper guide, the pelvic floor is the lower guide. release spasms and lengthen both.
Make room for the baby by releasing muscles that spasm, lengthen ligaments that are shortened, and support the abdominal muscles by attending to the muscles that interact with them, don’t go directly to the front first.
Featured Products
Shop spinning babies®.
For additional education to even further enhance your pregnancy and labor preparation, shop our extensive collection of digital downloads, videos, DVDs, workbooks, and more.

Connect with Us
Sign up for our newsletter:, more information:.
Have any questions or concerns? Email us at [email protected]
An expert resource for medical professionals Provided FREE as a service to women’s health
The Global Library of Women’s Medicine EXPERT – RELIABLE - FREE Over 20,000 resources for health professionals
The Alliance for Global Women’s Medicine A worldwide fellowship of health professionals working together to promote, advocate for and enhance the Welfare of Women everywhere

An Educational Platform for FIGO
The Global Library of Women’s Medicine Clinical guidance and resources
A vast range of expert online resources. A FREE and entirely CHARITABLE site to support women’s healthcare professionals
The Global Academy of Women’s Medicine Teaching, research and Diplomates Association
- Expert clinical guidance
- Safer motherhood
- Skills videos
- Clinical films
- Special textbooks
- Ambassadors
- Can you help us?
- Introduction
- Definitions
- Complications
- External Cephalic Version
- Management of Labor And Delivery
- Cesarean Delivery
- Perinatal Outcome
- Practice Recommendations
- Study Assessment – Optional
- Your Feedback
This chapter should be cited as follows: Okemo J, Gulavi E, et al , Glob. libr. women's med ., ISSN: 1756-2228; DOI 10.3843/GLOWM.414593
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Common obstetric conditions
Volume Editor: Professor Sikolia Wanyonyi , Aga Khan University Hospital, Nairobi, Kenya

Abnormal Lie/Presentation
First published: February 2021
Study Assessment Option
By completing 4 multiple-choice questions (randomly selected) after studying this chapter readers can qualify for Continuing Professional Development awards from FIGO plus a Study Completion Certificate from GLOWM See end of chapter for details

INTRODUCTION
The mechanism of labor and delivery, as well as the safety and efficacy, is determined by the specifics of the fetal and maternal pelvic relationship at the onset of labor. Normal labor occurs when regular and painful contractions cause progressive cervical dilatation and effacement, accompanied by descent and expulsion of the fetus. Abnormal labor involves any pattern deviating from that observed in the majority of women who have a spontaneous vaginal delivery and includes:
- Protraction disorders (slower than normal progress);
- Arrest disorders (complete cessation of progress).
Among the causes of abnormal labor is the disproportion between the presenting part of the fetus and the maternal pelvis, which rather than being a true disparity between fetal size and maternal pelvic dimensions, is usually due to a malposition or malpresentation of the fetus.
This chapter reviews how to define, diagnose, and manage the clinical impact of abnormalities of fetal lie and malpresentation with the most commonly occurring being the breech-presenting fetus.
DEFINITIONS
At the onset of labor, the position of the fetus in relation to the birth canal is critical to the route of delivery and, thus, should be determined early. Important relationships include fetal lie, presentation, attitude, and position .
Fetal lie describes the relationship of the fetal long axis to that of the mother. In more than 99% of labors at term, the fetal lie is longitudinal . A transverse lie is less frequent when the fetal and maternal axes may cross at a 90 ° angle, and predisposing factors include multiparity, placenta previa, hydramnios, and uterine anomalies. Occasionally, the fetal and maternal axes may cross at a 45 ° angle, forming an oblique lie .
Fetal presentation
The presenting part is the portion of the fetal body that is either foremost within the birth canal or in closest proximity to it. Thus, in longitudinal lie, the presenting part is either the fetal head or the breech, creating cephalic and breech presentations , respectively. The shoulder is the presenting part when the fetus lies with the long axis transversely.
Commonly the baby lies longitudinally with cephalic presentation. However, in some instances, a fetus may be in breech where the fetal buttocks are the presenting part. Breech fetuses are also referred to as malpresentations. Fetuses that are in a transverse lie may present the fetal back (or shoulders, as in the acromial presentation), small parts (arms and legs), or the umbilical cord (as in a funic presentation) to the pelvic inlet. When the fetal long axis is at an angle to the bony inlet, and no palpable fetal part generally is presenting, the fetus is likely in oblique lie. This lie usually is transitory and occurs during fetal conversion between other lies during labor.
The point of direction is the most dependent portion of the presenting part. In cephalic presentation in a well-flexed fetus, the occiput is the point of direction.
The fetal position refers to the location of the point of direction with reference to the four quadrants of the maternal outlet as viewed by the examiner. Thus, position may be right or left as well as anterior or posterior.
Unstable lie
Refers to the frequent changing of fetal lie and presentation in late pregnancy (usually refers to pregnancies >37 weeks).
Fetal position
Fetal position refers to the relationship of an arbitrarily chosen portion of the fetal presenting part to the right or left side of the birth canal. With each presentation there may be two positions – right or left. The fetal occiput, chin (mentum) and sacrum are the determining points in vertex, face, and breech presentations. Thus:
- left and right occipital presentations
- left and right mental presentations
- left and right sacral presentations.
Fetal attitude
The fetus instinctively forms an ovoid mass that corresponds to the shape of the uterine cavity towards the third trimester, a characteristic posture described as attitude or habitus. The fetus becomes folded upon itself to create a convex back, the head is flexed, and the chin is almost in contact with the chest. The thighs are flexed over the abdomen and the legs are bent at the knees. The arms are usually parallel to the sides or lie across the chest while the umbilical cord fills the space between the extremities. This posture is as a result of fetal growth and accommodation to the uterine cavity. It is possible that the fetal head can become progressively extended from the vertex to face presentation resulting in a change of fetal attitude from convex (flexed) to concave (extended) contour of the vertebral column.
The categories of frank, complete, and incomplete breech presentations differ in their varying relations between the lower extremities and buttocks (Figure 1). With a frank breech, lower extremities are flexed at the hips and extended at the knees, and thus the feet lie close to the head. With a complete breech, both hips are flexed, and one or both knees are also flexed. With an incomplete breech, one or both hips are extended. As a result, one or both feet or knees lie below the breech, such that a foot or knee is lowermost in the birth canal. A footling breech is an incomplete breech with one or both feet below the breech.

Types of breech presentation. Reproduced from WHO 2006, 1 with permission.
The relative incidence of differing fetal and pelvic relations varies with diagnostic and clinical approaches to care.
About 1 in 25 fetuses are breech at the onset of labor and about 1 in 100 are transverse or oblique, also referred to as non-axial. 2
With increasing gestational age, the prevalence of breech presentation decreases. In early pregnancy the fetus is highly mobile within a relatively large volume of amniotic fluid, therefore it is a common finding. The incidence of breech presentation is 20–25% of fetuses at <28 weeks, but only 7–16% at 32 weeks, and only 3–4% at term. 2 , 3
Face and brow presentation are uncommon. Their prevalence compared with other types of malpresentations are shown below. 4
- Occiput posterior – 1/19 deliveries;
- Breech – 1/33 deliveries;
- Face – 1/600–1/800 deliveries;
- Brow – 1/500–1/4000 deliveries;
- Transverse lie – 1/833 deliveries;
- Compound – 1/1500 deliveries.
Transverse lie is often unstable and fetuses in this lie early in pregnancy later convert to a cephalic or breech presentation.
The fetus has a relatively larger head than body during most of the late second and early third trimester, it therefore tends to spend much of its time in breech presentation or in a non-axial lie as it rotates back and forth between cephalic and breech presentations. The relatively large volume of amniotic fluid present facilitates this dynamic presentation.
Abnormal fetal lie is frequently seen in multifetal gestation, especially with the second twin. In women of grand parity, in whom relaxation of the abdominal and uterine musculature tends to occur, a transverse lie may be encountered. Prematurity and macrosomia are also predisposing factors. Distortion of the uterine cavity shape, such as that seen with leiomyomas, prior uterine surgery, or developmental anomalies (Mullerian fusion defects), predisposes to both abnormalities in fetal lie and malpresentations. The location of the placenta also plays a contributing role with fundal and cornual implantation being seen more frequently in breech presentation. Placenta previa is a well-described affiliate for both transverse lie and breech presentation.
Fetuses with congenital anomalies also present with abnormalities in either presentation or lie. It is possibly as a cause (i.e. fitting the uterine cavity optimally) or effect (the fetus with a neuromuscular condition that prevents the normal turning mechanism). The finding of an abnormal lie or malpresentation requires a thorough search for fetal abnormalities. Such abnormalities could include chromosomal (autosomal trisomy) and structural abnormalities (hydrocephalus), as well as syndromes of multiple effects (fetal alcohol syndrome).
In most cases, breech presentation appears to be as a chance occurrence; however, up to 15% may be owing to fetal, maternal, or placental abnormalities. It is commonly thought that a fetus with normal anatomy, activity, amniotic fluid volume, and placental location adopts the cephalic presentation near term because this position is the best fit for the intrauterine space, but if any of these variables is abnormal, then breech presentation is more likely.
Factors associated with breech presentation are shown in Table 1.
Risk factors for breech presentation.
Preterm gestation Previous breech presentation in sibling or parent Uterine abnormality (e.g., bicornuate or septate uterus, fibroid) Placental location (e.g., placenta previa Multiparity Extremes of amniotic fluid volume (polyhydramnios, oligohydramnios) Fetal anomaly (e.g., anencephaly, hydrocephaly, sacrococcygeal teratoma) Fetal neurologic impairment Fetal growth restriction Maternal anticonvulsant therapy Older maternal age Crowding from multiple gestation Extended fetal legs Short umbilical cord Contracted maternal pelvis Female sex |
Spontaneous version may occur at any time before delivery, even after 40 weeks of gestation. A prospective longitudinal study using serial ultrasound examinations reported the likelihood of spontaneous version to cephalic presentation after 36 weeks was 25%. 5
In population-based registries, the frequency of breech presentation in a second pregnancy was approximately 2% if the first pregnancy was not a breech presentation and approximately 9% if the first pregnancy was a breech presentation. After two consecutive pregnancies with breech presentation at delivery, the risk of another breech presentation was approximately 25% and this rose to 40% after three consecutive breech deliveries. 6 , 7
In addition, parents who themselves were delivered at term from breech presentation were twice as likely to have their offspring in breech presentation as parents who were delivered in cephalic presentation. This suggests a possible heritable component to fetal presentation. 8
Leopold’s maneuvers

The Leopold’s maneuvers: palpation of fetus in left occiput anterior position. Reproduced from World Health Organization, 2006, 1 with permission.
Abdominal examination can be conducted systematically employing the four maneuvers described by Leopold in 1894. 9 , 10 In obese patients, in polyhydramnios patients or those with anterior placenta, these maneuvers are difficult to perform and interpret.
The first maneuver is to assess the uterine fundus. This allows the identification of fetal lie and determination of which fetal pole, cephalic or podalic – occupies the fundus. In breech presentation, there is a sensation of a large, nodular mass, whereas the head feels hard and round and is more mobile.
The second maneuver is accomplished as the palms are placed on either side of the maternal abdomen, and gentle but deep pressure is exerted. On one side, a hard, resistant structure is felt – the back. On the other, numerous small, irregular, mobile parts are felt – the fetal extremities. By noting whether the back is directed anteriorly, transversely, or posteriorly, fetal orientation can be determined.
The third maneuver aids confirmation of fetal presentation. The thumb and fingers of one hand grasp the lower portion of the maternal abdomen just above the symphysis pubis. If the presenting part is not engaged, a movable mass will be felt, usually the head. The differentiation between head and breech is made as in the first maneuver.
The fourth maneuver helps determine the degree of descent. The examiner faces the mother’s feet, and the fingertips of both hands are positioned on either side of the presenting part. They exert inward pressure and then slide caudad along the axis of the pelvic inlet. In many instances, when the head has descended into the pelvis, the anterior shoulder or the space created by the neck may be differentiated readily from the hard head.
According to Lyndon-Rochelle et al ., 11 experienced clinicians have accurately identified fetal malpresentation using Leopold maneuvers with a high sensitivity 88%, specificity 94%, positive-predictive value 74%, and negative-predictive value 97%.
Vaginal examination
Prelabor diagnosis of fetal presentation is difficult as the presenting part cannot be palpated through a closed cervix. Once labor begins and the cervix dilates, and palpation through vaginal examination is possible. Vertex presentations and their positions are recognized by palpation of the various fetal sutures and fontanels, while face and breech presentations are identified by palpation of facial features or the fetal sacrum and perineum, respectively.
Sonography and radiology
Sonography is the gold standard for identifying fetal presentation. This can be done during antenatal period or intrapartum. In obese women or in women with muscular abdominal walls this is especially important. Compared with digital examinations, sonography for fetal head position determination during second stage labor is more accurate. 12 , 13
COMPLICATIONS
Adverse outcomes in malpresented fetuses are multifactorial. They could be due to either underlying conditions associated with breech presentation (e.g., congenital anomalies, intrauterine growth restriction, preterm birth) or trauma during delivery.
Neonates who were breech in utero are more at risk for mild deformations (e.g., frontal bossing, prominent occiput, upward slant and low-set ears), torticollis, and developmental dysplasia of the hip.
Other obstetric complications include prolapse of the umbilical cord, intrauterine infection, maldevelopment as a result of oligohydramnios, asphyxia, and birth trauma and all are concerns.
Birth trauma especially to the head and cervical spine, is a significant risk to both term and preterm infants who present breech. In cephalic presenting fetuses, the labor process prepares the head for delivery by causing molding which helps the fetus to adapt to the birth canal. Conversely, the after-coming head of the breech fetus must descend and deliver rapidly and without significant change in shape. Therefore, small alterations in the dimensions or shape of the maternal bony pelvis or the attitude of the fetal head may have grave consequences. This process poses greater risk to the preterm infant because of the relative size of the fetal head and body. Trauma to the head is not eliminated by cesarean section; both intracranial and cervical spine trauma may result from entrapment in either the uterine or abdominal incisions.
In resource-limited countries where ultrasound imaging, urgent cesarean delivery, and neonatal intensive care are not readily available, the maternal and perinatal mortality/morbidity associated with transverse lie in labor can be high. Uterine rupture from prolonged labor in a transverse lie is a major reason for maternal/perinatal mortality and morbidity.
EXTERNAL CEPHALIC VERSION
External cephalic version (ECV) is the manual rotation of the fetus from a non-cephalic to a cephalic presentation by manipulation through the maternal abdomen (Figure 3).

External version of breech presentation . Reproduced from WHO 2003 , 14 with permission .
This procedure is usually performed as an elective procedure in women who are not in labor at or near term to improve their chances of having a vaginal cephalic birth. ECV reduces the risk of non-cephalic presentation at birth by approximately 60% (relative risk [RR] 0.42, 95% CI 0.29–0.61) and reduces the risk of cesarean delivery by approximately 40% (RR 0.57, 95% CI 0.40–0.82). 7
In a 2008 systematic review of 84 studies including almost 13,000 version attempts at term, the pooled success rate was 58%. 15
A subsequent large series of 2614 ECV attempts over 18 years reported a success rate of 49% and provided more details): 16
- The success rate was 40% in nulliparous women and 64% in parous women.
- After successful ECV, 97% of fetuses remained cephalic at birth, 86% of which were delivered vaginally.
- Spontaneous version to a cephalic presentation occurred after 4.3% of failed attempts, and 2.2% of successfully vertexed cases reverted to breech.
Factors associated with lower ECV success rates include nulliparity, anterior placenta, lateral or cornual placenta, decreased amniotic fluid volume, low birth weight, obesity, posteriorly located fetal spine, frank breech presentation, ruptured membranes.
The following factors should be considered while managing malpresentations: type of malpresentation, gestational age at diagnosis, availability of skilled personnel, institutional resources and protocols and patient factors and preferences.
Breech presentation
According to a term breech trial, 17 planned cesarean delivery carries a reduced perinatal mortality and early neonatal morbidity for babies with breech presentation at term compared to vaginal breech delivery. When planning a breech vaginal birth, appropriate patient selection and skilled personnel in breech delivery are key in achieving good neonatal outcomes. In appropriately selected patients and skilled personnel in vaginal breech deliveries, perinatal mortality is between 0.8 and 1.7/1000 for planned vaginal breech birth and between 0 and 0.8/1000 for planned cesarean section. 18 , 19 The choice of the route of delivery should therefore be made considering the availability of skilled personnel in conducting breech vaginal delivery; providing competent newborn care; conducting rapid cesarean delivery should need arise and performing ECV if desired; availability of resources for continuous intrapartum fetal heart rate and labor monitoring; patient clinical features, preferences and values; and institutional policies, protocols and resources.
Four approaches to the management of breech presentation are shown in Figure 4: 8

Management of breech presentation. ECV, external cephalic version.
The options available are:
- Attempting external cephalic version (ECV) before labor with a trial of labor if successful and conducting cesarean delivery if unsuccessful.
- Footling or kneeling breech presentation;
- Fetal macrosomia;
- Fetal growth restriction;
- Hyperextended fetal neck in labor;
- Previous cesarean delivery;
- Unavailability of skilled personnel in breech delivery;
- Other contraindications to vaginal delivery like placenta previa, cord prolapse;
- Fetal anomaly that may interfere with vaginal delivery like hydrocephalus.
- Planned cesarean delivery without an attempt at ECV.
- Planned trial of vaginal breech delivery in patients with favorable clinical characteristics for vaginal delivery without an attempt at ECV.
All the four approaches should be discussed in detail with the patient, and in light of all the considerations highlighted above, a safe plan of care agreed upon by both the patient and the clinician in good time.
Transverse and oblique lie
If a diagnosis of transverse/oblique fetal lie is made before onset of labor and there are no contraindications to vaginal birth or ECV, ECV can be attempted at 37 weeks' gestation. If the malpresentation recurs, further attempts at ECV can be made at 38–39 weeks with induction of labor if successful.
ECV can also be attempted in early labor with intact fetal membranes and no contraindications to vaginal birth.
If ECV is declined or is unsuccessful, then planned cesarean section should be arranged after 39 weeks' gestation.
MANAGEMENT OF LABOR AND DELIVERY
Skills to conduct vaginal breech delivery are very important as there are women who may opt for planned vaginal breech birth and even among those who choose planned cesarean delivery, about 10% may go into labor and end up with a vaginal breech delivery. 17 Some implications of cesarean delivery such as need for repeat cesarean deliveries, placental attachment disorders and uterine rupture make vaginal birth more desirable to some individuals. In addition, vaginal birth has advantages such as affordability, quicker recovery, shorter hospital stay, less complications and is more favorable for resource poor settings.
In appropriately selected women, planned vaginal breech birth is not associated with any significant long-term neurological morbidity. Regardless of planned mode of birth, cerebral palsy occurs in approximately 1.5/1,000 breech births, and abnormal neurological development occurs in approximately 3/100. 18 Careful patient selection is very important for good outcomes and it is generally agreed that women who choose to undergo a trial of labor and vaginal breech delivery should be at low risk of complications from vaginal breech delivery. Some contraindications to vaginal breech delivery have been highlighted above.
Women with breech presentation near term, pre- or early-labor ultrasound should be performed to assess type of breech presentation, flexion of the fetal head and fetal growth. If a woman presents in labor and ultrasound is unavailable and has not recently been performed, cesarean section is recommended. Vaginal breech deliveries should only take place in a facility with ability and resources readily available for emergency cesarean delivery should the need arise.
Induction of labor may be considered in carefully selected low-risk women. Augmentation of labor is controversial as poor progress of labor may be a sign of cephalo-pelvic disproportion, however, it may be considered in the event of weak contractions. A cesarean delivery should be performed if there is poor progress of labor despite adequate contractions. Labor analgesia including epidural can be used as needed.
Vaginal breech delivery should be conducted in a facility that is able to carry out continuous electronic fetal heart rate monitoring sufficient personnel to monitor the progress of labor. From the term breech trial, 17 the commonest indications for cesarean section are poor progress of labor (50%) and fetal distress (29%). There is an increased risk of cord compression which causes variable decelerations. Since the fetal head is at the fundus where contractions begin, the incidence of early decelerations arising from head compression is also higher. Due to the irregular contour of the presenting part which presents a high risk of cord prolapse, immediate vaginal examination should be undertaken if membranes rupture to rule out cord prolapse. The frequency of cord prolapse is 1% with frank breech and more than 10% in footling breech. 8
Fetal blood sampling from the buttocks is not recommended. A passive second stage of up to 90 minutes before active pushing is acceptable to allow the breech to descend well into the pelvis. Once active pushing commences, delivery should be accomplished or imminent within 60 minutes. 18
During planned vaginal breech birth, a skilled clinician experienced in vaginal breech birth should supervise the first stage of labor and be present for the active second stage of labor and delivery. Staff required for rapid cesarean section and skilled neonatal resuscitation should be in-hospital during the active second stage of labor.
The optimum maternal position in second stage has not been extensively studied. Episiotomy should be undertaken as needed and only after the fetal anus is visible at the vulva. Breech extraction of the fetus should be avoided. The baby should be allowed to deliver spontaneously with maternal effort only and without any manipulations at least until the level of the umbilicus. A loop of the cord is then pulled to avoid cord compression. After this point, suprapubic pressure can be applied to facilitate flexion of the fetal head and descent.
Delay of arm delivery can be managed by sweeping them across the face and downwards towards in front of the chest or by holding the fetus at the hips or bony pelvis and performing a 180° rotation to deliver the first arm and shoulder and then in the opposite direction so that the other arm and shoulder can be delivered i.e., Lovset’s maneuver (Figure 5).

Lovset’s maneuver. Reproduced from WHO 2006 , 1 with permission .
The fetal head can deliver spontaneously or by the following maneuvers:
- Turning the body to the floor with application of suprapubic pressure to flex the head and neck.

Mauriceau-smellie-veit maneuver . Reproduced from WHO 2003, 14 with permission.
- By use of Piper’s forceps.
- Burns-Marshall maneuver where the baby’s legs and trunk are allowed to hang until the nape of the neck is visible at the mother’s perineum so that its weight exerts gentle downwards and backwards traction to promote flexion of the head. The fetal trunk is then swept in a wide arc over the maternal abdomen by grasping both the feet and maintaining gentle traction; the aftercoming head is slowly born in this process.
If the above methods fail to deliver the fetal head, symphysiotomy and zavanelli maneuver with cesarean section can be attempted. Duhrssen incisions where 1–3 full length incisions are made on an incompletely dilated cervix at the 6, 2 and 10 o’clock positions can be done especially in preterm.
Face presentation
The diagnosis of face presentation is made during vaginal examination where the presenting portion of the fetus is the fetal face between the orbital ridges and the chin. At diagnosis, 60% of all face presentations are mentum anterior, 26% are mentum posterior and 15% are mentum transverse. Since the submentobregmatic (face presentation) and suboccipitobregmatic (vertex presentation) have the same diameter of 9.5 cm, most face presentations can have a successful vaginal birth and not necessarily require cesarean section delivery. 6 The position of a fetus in face presentation helps in guiding the management plan. Over 75% of mentum anterior presentations will have a successful vaginal delivery, whereas it is impossible to have a vaginal birth in mentum posterior position unless it converts spontaneously to mentum anterior position. In mentum posterior position the neck is maximally extended and cannot extend further to deliver beneath the symphysis pubis (Figure 7).

Face presentation. Reproduced from WHO 2003, 14 with permission.
As in breech management, face presentation also requires continuous fetal heart rate monitoring, since abnormalities of fetal heart rate are more common. 5 , 6 In one study , 20 only 14% of pregnancies had normal tracings, 29% developed variable decelerations and 24% had late decelerations. Internal fetal heart rate monitoring with an electrode is not recommended, as it may cause facial and ophthalmic injuries if incorrectly placed. Labor augmentation and cesarean sections are performed as per standard obstetric indications. Vacuum and midforceps delivery should be avoided, but an outlet forceps delivery can be attempted. Attempts to manually convert the face to vertex or to rotate a posterior position to a more favorable anterior mentum position are rarely successful and are associated with high fetal morbidity and mortality, and maternal morbidity, including cord prolapse, uterine rupture, and fetal cervical spine injury with neurological impairment.
Brow presentation
The diagnosis of brow presentation is made during vaginal examination in second stage of labor where the presenting portion of the fetal head is between the orbital ridge and the anterior fontanel.
Brow presentation may be encountered early in labor, but is usually a transitional state and converts to a vertex presentation after the fetal neck flexes. Occasionally, further extension may occur resulting in a face presentation. The majority of brow presentations diagnosed early in labor convert to a more favorable presentation and deliver vaginally. Once brow presentation is confirmed, continuous fetal heart rate monitoring is necessary and labor progress should be monitored closely in order to pick any signs of abnormal labor. Since the brow diameter is large (13.5 cm), persistent brow presentation usually results in prolonged or arrested labor requiring a cesarean delivery. Labor augmentation and instrumental deliveries are therefore not recommended.
CESAREAN DELIVERY
This is an option for women with breech presentation at term to choose cesarean section as their preferred mode of delivery, for those with unsuccessful ECV who do not want to attempt vaginal breech delivery, have contraindications for vaginal breech delivery or in the event that there is no available skilled personnel to safely conduct a vaginal breech delivery. Women should be given enough and accurate information about pros and cons for both planned cesarean section and planned vaginal delivery to help them make an informed decision.
Since the publication of the term breech trial, 17 , 19 there has been a dramatic global shift from selective to planned cesarean delivery for women with breech presentation at term. This study revealed that planned cesarean section carried a reduced perinatal mortality and early neonatal morbidity for babies with breech presentation at term compared to planned vaginal birth (RR 0.33, 95% CI 0.19–0.56). The cesarean delivery rate for breech presentation is now about 70% in European countries, 95% in the United States and within 2 months of the study’s publication, there was a 50–80% increase in rates of cesarean section for breech presentation in The Netherlands.
A planned cesarean delivery should be scheduled at term between 39–41 weeks' gestation to allow maximum time for spontaneous cephalic version and minimize the risk of neonatal respiratory problems. 8 Physical exam and ultrasound should be performed immediately prior to the surgery to confirm the fetal presentation. A detailed consent should be obtained prior to surgery and should include both short- and long-term complications of cesarean section and the alternatives of care that are available. The abdominal and uterine incisions should be sufficiently large to facilitate easy delivery. Thereafter, extraction of the fetus is similar to what is detailed above for vaginal delivery.
Cesarean section for face presentation is indicated for persistent mentum posterior position, mentum transverse and some mentum anterior positions where there is standard indication for cesarean section.
Persistent brow presentation usually necessitates cesarean delivery due to the large presenting diameter that causes arrest or protracted labor.
Transverse/oblique lie
Cesarean section is indicated for patients who present in active labor, in those who decline ECV, following an unsuccessful ECV or in those with contraindications to vaginal birth.
For dorsosuperior (back up) transverse lie, a low transverse incision is made on the uterus and an attempt to grasp the fetal feet with footling breech extraction is made. If this does not succeed, a vertical incision is made to convert the hysterotomy into an inverted T incision.
Dorsoinferior (back down) transverse lie is more difficult to deliver since the fetal feet are hard to grasp. An attempt at intraabdominal version to cephalic or breech presentation can be done if membranes are intact before the uterine incision is made. Another option is to make a vertical uterine incision; however, the disadvantage of this is the risk of uterine rupture in subsequent pregnancies.
PERINATAL OUTCOME
Availability of skilled neonatal care at delivery is important for good perinatal outcomes to facilitate resuscitation if needed for all fetal malpresentations. 8 All newborns born from fetal malpresentations require a thorough examination to check for possible injuries resulting from birth or as the cause of the malpresentation.
Neonates who were in face presentation often have facial edema and bruising/ecchymosis from vaginal examinations that usually resolve within 24–48 hours of life and low Apgar scores. Trauma during labor may cause tracheal and laryngeal edema immediately after delivery, which can result in neonatal respiratory distress and difficulties in resuscitative efforts.
PRACTICE RECOMMENDATIONS
- Diagnosis of unstable lie is made when a varying fetal lie is found on repeated clinical examination in the last month of pregnancy.
- Consider external version to correct lie if not longitudinal.
- Consider ultrasound to exclude mechanical cause.
- Inform woman of need for prompt admission to hospital if membranes rupture or when labor starts.
- If spontaneous rupture of membranes occurs, perform vaginal examination to exclude the presence of a cord or malpresentation.
- If the lie is not longitudinal in labor and cannot be corrected perform cesarean section.
CONFLICTS OF INTEREST
Author(s) statement awaited.
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
1 | WHO. , 2nd edn. World Health Organization, 2006:51. Available: . |
2 | Scheer K, Nubar J. Variation of fetal presentation with gestational age. 1976;125(2):269–70. |
3 | Hickok DE, Gordon DC, Milberg JA, The frequency of breech presentation by gestational age at birth: a large population-based study. 1992;166(3):851–85 |
4 | Sorensen T, Hasch E, Lange AP. Fetal presentation during pregnancy. 1979;2(8140):477. |
5 | Hughey MJ. Fetal position during pregnancy. 1985;153(8):885–6. |
6 | Gardberg M, Leonova Y, Laakkonen E. Malpresentations–impact on mode of delivery. 2011;90(5):540–2. |
7 | Ghosh MK. Breech presentation: evolution of management. 2005;50(2):108–16. |
8 | Hofmeyr G. Overview of breech presentation. UpToDate [Internet] Waltham, MA: UpToDate. 2014. |
9 | Kastner I, Kachlik D. [German gynecologist and obstetrician Christian Gerhard Leopold (1846–1911)]. 2010;75(3):218–21. |
10 | Sharma JB. Evaluation of Sharma's modified Leopold's maneuvers: a new method for fetal palpation in late pregnancy. 2009;279(4):481–7. |
11 | Lydon-Rochelle M, Albers L, Gorwoda J, Accuracy of Leopold maneuvers in screening for malpresentation: a prospective study. 1993;20(3):132–5. |
12 | Ramphul M, Kennelly M, Murphy DJ. Establishing the accuracy and acceptability of abdominal ultrasound to define the foetal head position in the second stage of labour: a validation study. 2012;164(1):35–9. |
13 | Wiafe YA, Whitehead B, Venables H, The effectiveness of intrapartum ultrasonography in assessing cervical dilatation, head station and position: A systematic review and meta-analysis. 2016;24(4):222–32. |
14 | WHO. . Geneva: World Health Organization, 2003. Available: . |
15 | Grootscholten K, Kok M, Oei SG, External cephalic version-related risks: a meta-analysis. 2008;112(5):1143–51. |
16 | Melo P, Georgiou EX, Hedditch A, External cephalic version at term: a cohort study of 18 years' experience. 2019;126(4):493–9. |
17 | Hannah ME, Hannah WJ, Hewson SA, Planned caesarean section versus planned vaginal birth for breech presentation at term: a randomised multicentre trial. 2000;356(9239):1375–83. |
18 | Kotaska A, Menticoglou S. No. 384-Management of Breech Presentation at Term. 2019;41(8):1193–205. |
19 | No G-tG. management of breech presentation: green-top guideline No. 20b management of breech presentation: Green-top guideline No. 20b. 2017. |
20 | Benedetti TJ, Lowensohn RI, Truscott A. Face presentation at term. 1980;55(2):199–202. |
Online Study Assessment Option All readers who are qualified doctors or allied medical professionals can now automatically receive 2 Continuing Professional Development credits from FIGO plus a Study Completion Certificate from GLOWM for successfully answering 4 multiple choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about FIGO’s Continuing Professional Development awards programme CLICK HERE )
I wish to proceed with Study Assessment for this chapter
We use cookies to ensure you get the best experience from our website. By using the website or clicking OK we will assume you are happy to receive all cookies from us.
- The Open University
- Explore OpenLearn
- Get started
- Create a course
- Free courses
- Collections
My OpenLearn Create Profile
- Personalise your OpenLearn profile
- Save Your favourite content
- Get recognition for your learning
Already Registered?
- Antenatal Care Module: Ethiopian Federal Ministry of Health
- Labour and Delivery Care Module: Acknowledgements
- Labour and Delivery Care Module: Introduction
- Labour Delivery and Care Module: 1. Recognition of Normal Labour
- Labour and Delivery Care Module: 2. Assessing the Woman in Labour
- Labour Delivery and Care Module: 3. Care of the Woman in Labour
- Labour and Delivery Care Module: 4. Using the Partograph
- Labour and Delivery Care Module: 5. Conducting a Normal Delivery
- Labour and Delivery Care Module: 6. Active Management of the Third Stage of Labour
- Labour and Delivery Care Module: 7. Neonatal Resuscitation
- Introduction
- Learning Outcomes for Study Session 8
- 8.1.1 Vertex presentation
- 8.1.2 Malpresentations
- 8.1.3 Malposition
- 8.2 Causes and consequences of malpresentations and malpositions
- 8.3.1 Causes of breech presentation
- 8.3.2 Diagnosis of breech presentation
- 8.3.3 Types of breech presentation
- 8.3.4 Risks of breech presentation
- 8.4.1 Causes of face presentation
- 8.4.2 Diagnosis of face presentation
- 8.4.3 Complications of face presentation
8.5.1 Possible causes of brow presentation
8.5.2 Diagnosis of brow presentation
- 8.5.3 Complications of brow presentation
- 8.6.1 Causes of shoulder presentation
- 8.6.2 Diagnosis of shoulder presentation
- 8.6.3 Complications of shoulder presentation
- 8.7.1 Types of twin pregnancy
- 8.7.2 Diagnosis of twin pregnancy
- 8.7.3 Consequences of twin pregnancy
- 8.8 Management of women with malpresentation or multiple pregnancy
- Summary of Study Session 8
- Self-Assessment Questions (SAQs) for Study Session 8
- Labour and Delivery Care Module: 9. Obstructed Labour
- Labour and Delivery Care Module: 10. Ruptured Uterus
- Labour and Delivery Care Module: 11. Postpartum Haemorrhage
- Download PDF version
- Labour and Delivery Care PDF (4MB)
Download this course
Download this course for use offline or for other devices.
The materials below are provided for offline use for your convenience and are not tracked. If you wish to save your progress, please go through the online version.
About this course
- 26 hours study
- 1 Level 1: Introductory
- Course description

Labour and Delivery Care
If you create an account, you can set up a personal learning profile on the site.
You have seen all of these factors before, as causes of other malpresentations:
- Lax uterus due to repeated full term pregnancy
- Multiple pregnancy
- Polyhydramnios
- Abnormal shape of the mother’s pelvis.
8.5 Brow presentation
For further information, take a look at our frequently asked questions which may give you the support you need.
Have a question?
If you have any concerns about anything on this site please get in contact with us here.
Report a concern
What can the location of your headache really tell you? Experts explain
Plus a headache chart that can give you some idea what's going on

When it comes to aches and pains you don’t want to experience in your body, a headache is up there. But, while all headaches suck, they’re not created equal. There are actually several types of headaches, and they can show up in different areas of your head. That's why a headache chart can come in really handy.
Also, more common types of headaches can show up in a few different places: your sinus region (i.e., the center of your head and under your eyes), behind your eye, or on one side of your head. The reason? 'Headaches are often triggered, [and] the trigger typically drives the location of the headache,' says Amit Sachdev , MD, the director of the division of neuromuscular medicine at Michigan State University. 'For example, sinus and tension headaches tend to be the front of the face. Headaches associated with neck spasm tend to drive back of the head symptoms.'
Okay, so what are the different types of headaches and how can you figure out what's bothering you? Here’s everything you need to know.
Wait, so is the location of your headache important?
Again, where your headache is happening is and isn’t important. 'Location can play a role in determining the type of headache, but it’s not everything,' notes Jared Pomeroy , MD, MPH, a neurologist with Spectrum Health.
Different types of headaches can cause pain in the same area of your head, says Dr. Zhang, making location a tricky way to self-diagnose headaches.
Okay, got it. Then what are the most common types of headaches?
Headaches come in all kinds of shapes and sizes. In general, your headaches can be broken into the following groups.

Tension headache
A tension headache is the most common type of headache, according to the National Library of Medicine (NLM). It's usually brought on by stress or a result of muscle tension in your head, scalp, or neck. This type of headache usually makes you feel like a band is squeezing your head (fun!).
Tension headaches generally cause pain across the forehead or on both sides and the back of the head, according to the Mayo Clinic . They tend to come on slowly, and are usually mild to moderate.

Cluster headache
A cluster headache usually causes pain in and around one of your eyes or the side of your head, per the Mayo Clinic . It's considered rare compared to other types of headaches, and it's more common in men than women. It usually happens in phases called cluster periods and each attack generally lasts one to three hours, with the pain building up to a peak in 10 to 15 minutes
Besides the sudden onset of pain, you may also experience restlessness or agitation, red or watering eyes, a stuffy nose, sweating on the forehead, and eyelid drooping or swelling, according to Johns Hopkins Medicine .
The exact cause of cluster headaches isn’t known, but certain medications like nitroglycerin (a drug used to treat heart disease) can play a role, per the Mayo Clinic. 'This can be one of the worst headaches you have,' says Medhat Mikhael , MD, a pain management specialist and the medical director of the non-operative program at the Spine Health Center at MemorialCare Orange Coast Medical Center in Fountain Valley, California. 'It can feel like someone is poking a knife through your eye.'

Sinus headache
Sinus headaches generally feel like you’re having pain in your forehead, the bridge of your nose, or behind your cheekbones, according to the Cleveland Clinic . The pain usually gets worse when you move your head suddenly, and you may feel a constant dull ache.
This type of headache is a sign of a sinus infection, which causes pressure and pain in your face. You'll also have a fever, stuffy nose, thick and coloured mucus from the nose, feeling of fullness in the ears, and a puffy face. Anything from the common cold , seasonal allergies , nasal polyps, and a deviated septum could be to blame.
Heads up, though: Most of the time what people call a sinus headache is actually a migraine with nasal symptoms. Note that your discharge will be clear if you have a migraine.

Migraine headache
A migraine is a severe headache that usually causes pounding or throbbing pain on one side of your head. In addition to pain, they can also cause nausea and sensitivity to light, noise, or smell. Women are three times more likely than men to have migraines, per the Cleveland Clinic .
The cause of migraines is not fully understood, but they tend to run in families. You may also be more susceptible if you have a high stress level or are a smoker.
Migraines can be triggered by a slew of things, including stress, hormonal changes, light, certain foods and drinks, skipping a meal, caffeine, and lack of sleep, according to the Mayo Clinic .
Can COVID-19 cause headaches?
A headache is one of the main symptoms of COVID-19 , according to the Centers for Disease Control and Prevention (CDC), besides the usual fever, cough, sore throat, and muscle or body aches.
'Headache is very common in COVID-19 infections,' Dr. Sachdev says. 'These typically look like tension style headaches with the forehead or face aching.'
To get relief, Dr. Mikhael recommends using an anti-inflammatory medication like acetaminophen or ibuprofen. Adequate hydration and some caffeinated products can also help, he adds.
When should you worry about your headache?
Dr. Sachdev recommends keeping an eye out for 'red flag' symptoms that indicate you should seek treatment soon, such as:
- Systemic symptoms like a fever or night sweats
- Weakness that’s new
- Numbness that’s new
- A 'thunderclap' headache that comes out of nowhere and feels like the worst headache of your life
You’ll also want to see your doctor if you’re having regular headaches that cause you to treat it with over-the-counter medication more than four days a month, says Dr. Pomeroy. 'If your headache is affecting your work or having a significant impact on your life, you really owe it to yourself to speak to your physician,' he adds.

Read now: 5 things to know before you get a bob
More from Women's Health...
- How to stop "worry-wasting" your time
- 5 ways to deal with chin acne, and what causes it
- 5 signs of high oestrogen and what could be causing it
Cut through the noise and get practical, expert advice, home workouts, easy nutrition and more direct to your inbox. Sign up to the WOMEN'S HEALTH NEWSLETTER
'I used a free sperm donor from Facebook'

‘We need to call these riots what they are’

New drug reduces menopausal osteoporosis approved

Your face could use a massage—here’s why

Experts say this is how to stop people pleasing

Problematic smartphone use linked to teen anxiety

How to stop worry-wasting your time

The patients who DIY-d cluster headache treatment

‘The real ADHD scandal is NHS underfunding’

What are the early signs of a thyroid problem?

How box breathing can soothe you during stress
Cystic acne can cause pain, shame and lasting scars. Here's what causes it.

Dealing with cystic acne can be painful and scarring — physically and mentally.
Cystic acne gets its name from cysts, which are pus-filled pimples . They're typically large, painful and set deep in the skin, which often leads them to leave scars in the aftermath, according to the American Academy of Dermatology Association (AAD)
Acne is the most common skin condition in the United States and affects upwards of 50 million Americans every year, per the AAD. But cystic acne in particular can often be a source of embarrassment and anxiety for those who are dealing with it.
"Please know that acne is very normal and it's not your fault, and you are in excellent company — about 90% of people struggle with acne at some point in their life, and this includes celebrities," board-certified dermatologist Hadley King, M.D. , tells USA TODAY. "Acne, unfortunately, is normal and largely out of our control and it does not define who we are."
Here's what skin experts want you to know about cystic acne.
What causes cystic acne?
Cystic acne typically arises for people in their teens and 20s, but it can last into adulthood as well. The cause is usually hormonal, Dr. King says, with the caveat that it doesn't necessarily mean there's something wrong with your hormones.
"They result from the normal fluctuation of hormones," she says. Some people's sebaceous glands, which produce oil to prevent the skin from drying out, are genetically more sensitive to these hormones, causing more acne when those levels fluctuate. Things like stress, diet and lack of sleep can also impact hormones and acne.
More: TikTokers are using blue light to cure acne. Dermatologists say it's actually a good idea.
How can I prevent cystic acne?
Because cystic acne is usually caused by hormones, King notes that treatment is often the same for cystic and hormonal acne.
Daily cleansing is step No. 1 when it comes to any type of acne. It's best to consult a dermatologist to determine what else would be helpful, be it a tretinoin, retinoid or benzoyl peroxide. If those don't help, your dermatologist may also suggest some other treatment options for hormonal acne:
- Clascoterone , a newer topical cream, is the first FDA-approved hormonal acne medication for men and women. Medical experts believe it works by blocking your skin's hormones from making too much sebum, which can clog pores and cause acne.
- Spironolactone , an oral medication prescribed to women that is commonly prescribed along with oral contraceptives to address hormonal acne, per the AAD. "For women who have stubborn hormonal acne, this medication can effectively treat acne on the face, chest and back," the AAD notes.
- Isotretinoin , an oral retinoid, can also be helpful for cystic acne, King says.
Your browser is unsupported
We recommend using the latest version of IE11, Edge, Chrome, Firefox or Safari.
Drug Information Group
College of pharmacy - chicago | rockford, what is the recommended dosage of anakinra (kineret) for secondary hemophagocytic lymphohistiocytosis (hlh), text 1 heading link copy link.
Introduction Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome of excessive immune activation that can be life-threatening. 1,2 Most commonly, HLH impacts infants, but is also observed in children and adults. Diagnosis of HLH can be challenging, as there is a large variation in presentation and severity. HLH may typically present as an acute illness with accompanying fever associated with multiple organ involvement, and initial signs and symptoms can present like common infections, complicating diagnosis. In primary HLH, etiology is thought to be due to genetic factors and most often due to autosomal recessive genetic mutations. Secondary HLH is described as acquired, often resulting from infections, malignancies, or autoimmune diseases. Macrophage-activation syndrome-associated HLH is another term used for autoimmune disease-induced HLH. Classification may not fall neatly into primary or secondary HLH, as there are many overlapping risk factors, and infection can be a trigger for both. Persistent activation of inflammatory cell types, including macrophages, natural killer cells, and cytotoxic lymphocytes can lead to excess cytokine release, creating a cytokine storm, which is likely responsible for multiorgan failure in individuals with HLH.
The overall frequency of HLH, while unknown, is thought to be rare. 1,2 However, secondary HLH is associated with a very high acute all-cause mortality of approximately 40% in adults, rising to as high as 85% when incited by malignancy. Supportive intensive care is often required to treat patients with secondary HLH or macrophage-activation syndrome-HLH. Identification of underlying triggers and prompt treatment directed at these triggers is critical. Additional immunosuppressive or immunomodulatory HLH-directed therapy may also be warranted.
Anakinra has been used as HLH-directed therapy for the treatment of secondary HLH or macrophage-activation syndrome-HLH, yet specific dosing recommendations are lacking. 1 The purpose of this article is to describe the literature and associated dosage of anakinra in patients with secondary HLH and macrophage-activation syndrome-HLH.
Anakinra in HLH Anakinra (Kineret) is a disease-modifying antirheumatic, interleukin-1 (IL-1) receptor antagonist. 3 It is US Food and Drug Administration (FDA) approved for use in rheumatoid arthritis, cryopyrin-associated periodic syndromes, and deficiency of IL-1 receptor antagonist. Its use in secondary HLH is an off-label indication. 1,2 Since IL-1 is central to cytokine storm syndrome in patients with HLH, anakinra is thought to target and terminate hyperinflammation associated with IL-1 in HLH. The evidence evaluating anakinra in patients with secondary HLH or macrophage-activation syndrome is limited to several case reports/series and a systematic review of case reports.
Systematic reviews Charlesworth and Kavirayani (2023) identified 29 case reports/series with 87 patients (median age, 22 years; range, 22 months to 84 years) with secondary HLH who received intravenous (IV) anakinra as part of treatment. 4 The goal of the narrative systematic review was to collate data on the use of IV anakinra in these patients. Many of the underlying triggers precipitating secondary HLH or macrophage-activation syndrome-HLH were identifiable, and included significant infection (43.2%), rheumatological causes (33.3%), oncological causes (8.6%), or another known trigger (14.8%). Adverse reactions attributed to anakinra were reported in 9 cases (10.3%), most of which were minor. The overall survival was 67.8% of the 87 cases reported, but the timing reporting survival varied from 1 to 2 weeks following anakinra treatment to full recovery after many years. The pediatric patients had a greater overall survival (77%) compared to adult patient (61.5%). Of the cases reported, the administered dose of IV anakinra was available for 77.9% of cases. All doses were normalized to mg/kg/day to allow dose comparisons. After normalization, the daily dose of IV anakinra ranged from 1.7 to 48 mg/kg/day (median, 6.86 mg/kg/day). Anakinra dosing was significantly higher in younger patients, but this relationship was only significant amongst patient with infection triggers, when examined by underlying cause. For infection-precipitated HLH, the anakinra dose was significantly higher with reducing age. Most patients received IV bolus dosing (n=75, 87.2%), with some (n=11) reported as receiving continuous IV infusion.
Case series Numerous additional retrospective case studies and chart review studies have evaluated anakinra dosing in patients with secondary HLH. 5-8 Identified large case series (2 or more patients) not included in the above systematic review are summarized in the Table.
table 1 Heading link Copy link
| Miettunen 2011 Retrospective case series Patients followed for a median of 22 (range, 2 to 40) months | N=12 pediatric patients with rheumatic disease-associated MAS-HLH (age range, 6 months to 17 years) | Anakinra 2 mg/kg/day (max dose, 100 mg/day) once daily, subcutaneously All patients initially received corticosteroids and other immunosuppressants with limited benefit | Median hospitalization stay before anakinra was 11 (range, 1 to 27) days. All patients achieved MAS remission after addition of anakinra within a median of 13 (range, 2 to 19) days, and were in remission at the final follow-up | Anakinra 2 mg/kg/day once daily subcutaneously, combined with other immunosuppressants, led to remission of MAS-HLH in 12 pediatric patients with underlying rheumatic disease |
| Wohlfarth 2017 Retrospective case series Length of ICU stay (range, 3 to 118 days) | N=8 adult patients with HLH who received ICU treatment (median age, 38 years; range, 20 to 58 years) 63% (n=5) had a precipitating factor identified | Anakinra absolute dose of 100 to 200 mg three times daily, subcutaneously (median daily dose per kg body weight, 6 mg; range, 4 to 8 mg/kg/day) All patients initially received corticosteroids and IVIG with limited benefit | Median duration of anakinra administration in ICU survivors was 18 (range, 7 to 42) days | Anakinra combined with IVIG and/or corticosteroids resulted in a 50% survival rate in 8 critically ill adults with secondary HLH |
| Bami 2020 Retrospective chart review Follow-up, 3.5 to 5.5 years | N=6 pediatric patients with secondary HLH (median age, 1.8 years; range, 0.8 to 14.9 years) | Initial treatment with anakinra 6 to 10 mg/kg/day divided over 4 doses, subcutaneously 5 of the 6 children were also treated with dexamethasone | Average treatment duration was 8 weeks; 3-year overall survival was 83% (n=5/6) No patient required escalation of therapy to etoposide and all patients achieved remission. Anakinra was well tolerated without significant adverse events | Anakinra as a first-line treatment is feasible for treatment of pediatric secondary HLH |
| Baverez 2022 Retrospective chart review Median follow-up, 10 months | N=21 patients with secondary HLH (median age, 45 years) | Anakinra subcutaneously, at doses of 100 mg/day (n=14), 200 mg/day (n=5), or 2 to 5 mg/kg/day in children (n=3) Used as monotherapy in 5 patients, in combination with corticosteroids as initial treatment in 5 patients, and as second-line or beyond in 11 patients | 19/21 patients (90.5%) experienced remission after anakinra. In 3 of these patients, the dosage was doubled to achieve remission Fever resolved in 19 patients within a median of 1 day | Anakinra, alone or in combination with corticosteroids, led to favorable outcomes for patients with secondary HLH |
| Abbreviations: HLH=hemophagocytic lymphohistiocytosis; ICU=intensive care unit; IVIG=IV immune globulin; MAS=macrophage-activated syndrome. | ||||
text 2 Heading link Copy link
Relevant guidelines In 2022, the Histiocyte Society published a consensus-based guideline for the recognition, diagnosis, and management of HLH in critically ill children and adults, which was endorsed by the Society of Critical Care Medicine. 2 Within the guidelines, the authors provided recommended therapies and dosages based on available case reports of varying HLH severity. as summarized in the Table below.
table 2 Heading link Copy link
| Secondary HLH | Mild | Consider addition of corticosteroid therapy |
| Moderate | Dexamethasone 10 mg/m daily, divided every 12 hours, or equivalent methylprednisolone dosing (2 mg/kg/d) Consider addition of anakinra 2 to 10 mg/kg/d, divided in 2 to 4 daily doses (subcutaneous or IV) | |
| Severe, progressive, or refractory | Addition of etoposide with dose reduction as follows: 100 mg/m once weekly in older teens 75 mg/m once weekly in adults 50 mg/m once weekly in the elderly Renal dose reduction is recommended; dose reduction for hypoalbuminemia, hyperbilirubinemia alone, other evidence of liver dysfunction, and/or cytopenias is not recommended | |
| Macrophage-activation syndrome-HLH | Mild | Corticosteroids (such as methylprednisolone 30 mg/kg/d with maximum 1 g/d, for 3 to 5 days) with or without IVIG |
| Moderate | Consider addition of anakinra (dosing as above) and/or cautiously dosed cyclosporine (2 mg/kg/d in 2 divided doses, with a goal of serum levels 100 to 150 ng/mL) and/or consideration of tocilizumab | |
| Severe, progressive, or refractory | Consider addition of etoposide or cyclophosphamide | |
| Abbreviations: HLH=hemophagocytic lymphohistiocytosis; IV=intravenous; IVIG=IV immune globulin; pSOFA=pediatric sequential organ failure assessment; SOFA=sequential organ failure assessment. Mild defined as no evidence of organ dysfunction except coagulation/hematologic system; Moderate defined as evidence of moderate organ dysfunction (SOFA or pSOFA score 2 or less per organ system excluding coagulation/hematologic system) and possible need for supplemental oxygen; Severe defined as evidence of severe organ dysfunction (SOFA or pSOFA score 3 or more of at least 1 organ system excluding coagulation/hematologic system) and/or any need for organ replacement therapy due to organ failure, including positive-pressure ventilation, renal replacement therapy, vasopressors, and extracorporeal life support. | ||
text 3 Heading link Copy link
Conclusion Numerous case series highlight the efficacy of anakinra as monotherapy, or in combination with other immunosuppressants, as initial or second-line and beyond therapy for individuals with secondary HLH. Consensus-based guidelines from the Histiocyte Society recommend anakinra 2 to 10 mg/kg/day, in 2 to 4 daily doses, administered subcutaneously or IV. In general, case reports and systematic reviews of case reports support this dosing strategy, although some report even higher doses in certain patients. To appropriately determine anakinra’s place in therapy, prospective studies are needed comparing anakinra to other recommended treatment options, yet, due to the life-threatening nature of HLH, anakinra remains a viable option in this patient population.
- Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020;2(6): e358-e367. doi: 10.1016/S2665-9913(20)30096-5
- Hines MR, von Bahr Greenwood T, Beutel G, et al. Consensus-based guidelines for the recognition, diagnosis, and management of hemophagocytic lymphohistiocytosis in critically ill children and adults. Crit Care Med. 2022;50(5):860-872. doi: 10.1097/CCM.0000000000005361
- Anakinra. Package insert. Biovitrum AB; 2020.
- Charlesworth JEG, Kavirayani A. Intravenous anakinra for the treatment of haemophagocytic lymphohistiocytosis/macrophage activation syndrome: a systematic review. Eur J Haematol. 2023;111(3):458-476. doi: 10.1111/ejh.14029
- Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford). 2011;50(2):417-417. doi: 10.1093/rheumatology/keq218
- Wohlfarth P, Agis H, Gualdoni GA, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically ill adults with hemophagocytic lymphohistiocytosis. J Intensive Care Med. 2019;34(9):723-731. doi: 10.1177/0885066617711386
- Bami S, Vagrecha A, Soberman D, et al. The use of anakinra in the treatment of secondary hemophagocytic lymphhistiocytosis. Pediatr Blood Cancer. 2020;67(11):e28581. doi: 10.1002/pbc.28581
- Baverez C, Grall M, Gerfaud-Valentin M, et al. Anakinra for the treatment of hemophagocytic lymphohistiocytosis: 21 cases. J Clin Med. 2022;11(19):5799. doi: 10.3390/jcm11195799
Prepared by: Rachel Brunner, PharmD, BCPS Clinical Assistant Professor, Drug Information Specialist University of Illinois Chicago College of Pharmacy
August 2024
The information presented is current as of July 12, 2024. This information is intended as an educational piece and should not be used as the sole source for clinical decision-making.
- Case Report
- Open access
- Published: 10 August 2024
Chrysosporium articulatum mimicking Trichophyton spp. infection in a cat: a case presentation and literature review
- Magdalena Kizerwetter-Świda 1 ,
- Iwona Bąk 1 ,
- Małgorzata Justyna Biegańska 1 ,
- Kourou Dembele 2 &
- Dorota Chrobak-Chmiel 1
BMC Veterinary Research volume 20 , Article number: 359 ( 2024 ) Cite this article
Metrics details
Dermatophytosis is a common skin infection of cats and many other animals. A reliable diagnosis is crucial because of the zoonotic potential of dermatophytes. The routine mycological diagnostic procedures for dermatophytosis are widely known, but in the case of some isolates, identification based on phenotypic characteristics may be incorrect. Infections caused by Chrysosporium spp. are usually described in reptiles, but in other animals they are uncommon.
Case presentation
This study presents a description of a cat with dermatological lesions, that was mistakenly diagnosed with Trichophyton spp. dermatophytosis. Clinical material for mycological examination was collected from alopecic areas on the back of the neck, the ventral abdomen, and the hindlimbs. The initial identification based on phenotypic properties indicated Trichophyton spp. The result of the MALDI-ToF MS allowed the exclusion of the Trichophyton genus. Ultimately, the correct identification as Chrysosporium articulatum was obtained based on the sequencing of ribosomal genes.
Conclusions
Interpretation of the results of the mycological examination of samples collected from animals’ skin or hair shafts is always challenging. Thus, careful consideration of the primary cause of the clinical lesions observed on the skin is mandatory, and the culture results are worth supporting by molecular methods.
Peer Review reports
Dermatophytosis is a common fungal infection in veterinary and human medicine. Dermatophytes are filamentous fungi that may cause superficial infections of keratinized tissues such as skin (stratum corneum of the epidermis), hairs and claws in different animal species, including dogs and cats. The vast majority of dermatophytoses in pets are caused by Micropsorum spp. and Trichophyton spp. [ 1 , 2 , 3 , 4 ]. The pathogenicity of these fungi is related to their ability to degrade keratin found in superficial tissues, typically viable tissues are rather not invaded. However, sporadic invasive infections have been reported in immunocompromised or elderly human patients [ 5 ]. Dermatophytes belong to a group of keratinophilic and keratinolytic fungi. In addition, many keratinophilic environmental fungal species can use pre-digested keratinaceous debris or by-products of keratin degradation. These are: Chrysosporium spp., Psuedogymnoascus spp., Geomyces spp., Pectinotrichum spp., Renispora spp. and others. In general, these non-dermatophytes keratinolytic fungi are saprophytes, engaged in the decomposition of keratinized residues in the soil. However, Chrysposporium spp. strains with kertinolytic properties have been described, with positive results in hair perforation test [ 6 , 7 ].
Chrysosporium genus is classified in the family Onygenaceae , Onygenales order, Eurotiomycetes class and Ascomycota phylum. This genus includes about 100 species [ 8 ], commonly found in the environment, soil, and water sediments, but also on the skin and hairs of animals and humans. The taxonomical classification is often based on the fungal morphology. However, when sexual states and macroconidia are not present, the microconidia-producing fungi are clustered in polyphyletic genera, such as the genus Chrysosporium . Recent research results based on genetic properties have allowed the updating of the Chryspsporium spp. taxonomy [ 9 ]. Moreover, Kendemir et al. (2022) have shown 100% ITS sequence identity in C. articulatum UAMH 4320 with Aphanoascus reticulisporus [ 10 ]. Colonies formed by Chrysosporium are white or pale with septate hyphae producing pyriform or obovate to ellipsoidal microconidia [ 6 ]. The appearance of these powdery colonies as well as micromorphology resembles some dermatophytes, e.g. Trichophyton mentagrophytes [ 11 ]. Fungi classified in the genus Chrysosporium are regarded as non-pathogenic. However, there has been an increasing number of infections caused by these fungi in recent years. Most of the documented cases involve immunocompromised humans [ 12 , 13 ]. Infections of this etiology also occur in animals, mainly in reptiles, most often as cases of dermatitis, but also as life-treating infections [ 14 , 15 ]. Chrysosporium tropicum was described as a causative agent for dermatomycosis in chickens [ 16 ]. Additionally, Chrysosporium spp. is often isolated from feathers [ 17 ].
The clinical manifestations of dermatophytosis in cats are variable and related to the dermatophyte species involved [ 18 ]. Typically, single or several alopecic areas with scaling, crusting and erythema are observed. However, other clinical presentations are also possible, like a matted coat, seborrhea, miliary dermatitis, the presence of pustules, papules, macules, nodules, hyperpigmentation, kerions, and onychomycosis. Infected animals may show symptoms of pruritus. The variable clinical appearance of dermatophytosis can be explained by differences in the composition and structure of keratin, the specificity of enzymes produced by particular fungi, and the defence mechanisms of host organisms [ 18 , 19 ]. Moreover, any other dermatoses may cause similar clinical manifestations. Thus, differential diagnosis including, among others food allergy, hormonal disorders, atopic dermatitis, autoimmune dermatoses, bacterial dermatitis, or infestation with skin parasites should always be performed. Hence, the diagnostic procedures must be accurate and carried out step-by-step. Apart from mycological examination, the results of additional tests such as parasitic, bacteriological, histopathology of biopsy material and allergy tests should also be performed [ 19 ]. Of note, the reliable diagnosis of dermatophytosis in dogs and cats is also essential because of the zoonotic potential of most of the species isolated from pets [ 20 ]. Moreover, cats may be asymptomatic carriers of M. canis or they may have a subclinical infection, which further complicates the diagnosis [ 3 ].
In this study we present a case of a cat with dermatological lesions, initially diagnosed with Trichophyton spp. infection. Ultimately, the cultured fungi were identified by sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry method (MALDI-ToF MS) as C. articulatum , which is usually regarded as a non-pathogenic fungus. Moreover, we present a review of diagnostic procedures used in dermatophyte identification and the literature data on infections caused by Chrysosporium spp.
A 7-year-old an outdoor, neutralized male European shorthair cat weighing 6 kg showing dermatological lesions was admitted to the Small Animal Clinic at the Institute of Veterinary Medicine, Warsaw University of Life Sciences. Clinical findings included: intense pruritus and alopecia on the back of the neck, on the ventral abdomen, and the hindlimbs (Fig. 1 ). At the visit, flea dermatitis was excluded. Wood’s lamp examination was performed, and no fluorescence was observed. The cat was diagnosed with dermatitis miliaris. To reduce intense itching, the cat was treated with dexafort (0,9 mg i.m.). Plucked hairs and scraped scales were collected for mycological examination.

Pruritic self-inflicted alopecic areas on the back of the neck (left) and the hindlimb (right)
Direct microscopic examination of collected hairs and scales was performed with KOH, but wet-mounts failed to detect any spores or other fungal elements in both examined samples. Sabouraud dextrose agar (SDA), Sabouraud dextrose agar supplemented with 0.05% cycloheximide and 0.005% chloramphenicol, and dermatophyte test medium (DTM) were used for fungal culture. All plates were incubated aerobically, at 25 °C for four weeks. The colonies appeared on SDA and DTM medium after five days of incubation. Colonies were flat, white in colour, with a powdery surface (Fig. 2 ). DTM medium turned red, as is observed when dermatophytes grow. Colony morphology resembled colonies of Trichophyton spp. (Fig. 3 ). The isolate was examined for microscopic morphology using lactophenol cotton blue staining. Conidia were smooth and thin-walled, pyriform, one-celled, and sessile, usually on side branches or at the ends of long narrow stalks (Fig. 4 ). Additionally, a hair perforation test was performed following standard mycological procedures, and no keratinolysis was noted. The isolate was identified based on the colony morphology on SDA, DTM medium and micromorphology as Trichophyton spp. Thus, topical and systematic antifungal therapy was prescribed.
The fungal isolate was further identified using MALDI Biotyper (Bruker Daltonics, Billerica, MA, USA) according to the manufacturer’s instruction at the Jagiellonian Centre of Innovation (Kraków, Poland). The identification of our isolate with the MALDI-ToF MS method revealed Chrysosporium keratinophilum with a score value of 2.11. The identification score ranging 2.00–3.00 was considered as a high-confidence identification to the species level.
Ultimately, molecular biology methods were used for identification. Genomic DNA was extracted from five-day-old colonies using the method described by Brillowska-Dabrowska et al. [ 21 ]. Briefly, a fragment of a colony was mixed with 100 µl of extraction buffer (60 mM sodium bicarbonate, 250 mM potassium chloride and 50 mM Tris, pH 9.5, Sigma Aldrich) and incubated at 95 °C for 10 min. Then, 100 µl of 2% bovine serum albumin was added and after vigorous vortexing for 5 s, the obtained solution was used for PCR. Amplification of the internal transcribed sequence (ITS) region of ribosomal RNA was used with conserved primers ITS4 and ITS5 described by White et at. [ 22 ], with the following thermal-cycling conditions: initial denaturation for 3 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 50 °C, 45 s of at 72 °C, and final elongation for 6 min. The obtained product was verified by agarose gel electrophoresis and subjected to sequencing with the same primers. Finally, the sequence was analyzed with BLAST software using the National Center for Biotechnology Information (NCBI) database. GenBank BLAST analysis of the obtained sequence of the internal transcribed sequence region of ribosomal RNA indicated 99.27% identity to a sequence of Chrysosporium articulatum deposited in the NCBI database.
Finally, the isolate obtained from a cat was recognized as C. articulatum , which was considered an environmental isolate contaminating the fur. Based on the verified identification dermatophytosis was ultimately excluded, allowing to avoid unnecessary implementation of antifungal therapy to the patient. The final diagnosis was a food allergy, with the recommendation of an elimination diet. After four weeks, a follow-up visit took place, during which the veterinarian confirmed that the cat’s condition improved, in alopecic areas, fur started to regrow and the itching had stopped. During the follow-up visit, hair samples were collected for mycological culture, which gave a negative result.

Colony morphology on SDA medium supplemented with chloramphenicol and cycloheximide (front and back of the plate) - flat, white colonies, with a powdery surface on the front and pale brown on the reverse

Colony morphology on DTM medium - colour change from yellow to red (five days of incubation on the left and four weeks of incubation on the right)

Morphology of septate hyphae and microconidia – light microscope examination under 400x magnification with lactophenol cotton blue staining
Veterinary mycological diagnostics encounter certain difficulties in identifying unusual, less frequently isolated species. The positive fungal culture results in invasive infections or disseminated cutaneous infections and does not pose any problems in interpretation because the clinical samples are collected from tissues and should not contain any fungal elements, including saprophytes. The cultivation of fungi commonly considered environmental saprophytes from superficial skin lesions is more challenging in interpretation. It may be difficult to assess whether these fungi caused the infection (in some immunocompromised patients) or whether they were cultivated accidentally. Moreover, in some cases, the differentiation of dermatophytes and other non-dermatophytic fungi may be more demanding than it seems. Incorrect identification of pathogenic fungi as saprophytes may result in the omitting of necessary antifungal therapy despite the medical indications. Alternatively, therapy may be introduced for patients that do not require such treatment, because only environmental saprophytic fungi were cultured from samples collected superficially. The treatment of dermatophytosis in dogs and cats may be topical or quite often requires systemic administration of antifungals [ 23 ]. Topical therapy is used to minimize disease transmission and environmental contamination, while systemic antifungal therapy eradicates the infection within the hair follicle [ 24 ]. Treatment of dermatophytosis may be associated with side effects, such as liver toxicity or vasculitis, and it may lead to an increase in fungal resistance. Unnecessary antifungal treatment, which is usually long-term, causes an imbalance in natural microbiota.
Fungi classified in the genus Chrysosporium are regarded as non-pathogenic, non-dermatophyte keratinolytic fungi. Recently, the number of cases of human infections caused by Chrysosporium spp. described in the literature is increasing, especially in immunocompromised human patients. Chrysosporium zonatum and Chrysosporium tropicanum are most commonly reported [ 25 ]. The clinical presentation includes respiratory allergic reactions, pulmonary invasive infections and skin infections. There is only one documented case of Chrysosporium articulatum invasive pulmonary infection in human, 16-year-old man diagnosed with lymphoblastic leukemia [ 12 ].
In veterinary medicine infections caused by Chrysosporium spp. are rarely described, and mostly are reported in reptiles. In recent years, Chrysosporium anamorph of Nannizziopsis vriesii (CANV) has become the leading fungal agent of dermatitis in reptiles. The lesions initially involve the skin, and the presence of hyperkeratosis, necrosis, vesicles, crusts, and ulceration may be observed. Progress to fatal systemic disease often occurs [ 14 , 15 ].
We have gathered here five literature reports concerning Chrysosporium spp. infections in dogs and cats. Of note, publications describing the isolation of these fungi from before 1990 have been omitted due to the unreliable identification methods used at that time. The first is a review study concerning 157 cases of disseminated canine mould infections demonstrated that the majority (59,3%) was caused by Aspergillus spp. Chrysosporium spp. was identified as the etiological agent only in two cases, which corresponds to only 1,3% of incidence [ 26 ]. One of the publications included in the review mentioned above was a case report concerning disseminated infection in German shepherd dog in Australia. Fungal hyphae were observed in needle aspirates of the iliac lymph nodes and spleen. The fungal culture from these materials was positive and was diagnosed as Chrysosporium spp. [ 27 ]. An earlier publication also from Australia described disseminated opportunistic fungal infections among 10 dogs, of which, in one case, Chrysosporium spp. was found to be the etiological agent [ 28 ]. In another review study describing fungal keratitis in 11 dogs, the presence of Chrysosporium spp. was confirmed in one patient [ 29 ]. Moreover, the literature provides one description of superficial skin lesions in two Persian cats and their owner caused by Chrysosporium spp. These two cats lived in the same household. Moreover, Chrysosporium spp. was also isolated from its owner, who was undergoing chemotherapy for mammary cancer. Fungal culture from hairs and skin scrapings revealed the presence of Chrysosporium spp. in both cats. Unfortunately, the authors did not verify the identification with molecular biology methods, however, effective antifungal treatment proved, that the isolated fungi were the etiological agent involved in the observed clinical changes [ 30 ]. Additionally, in 2011 Pin et al. described well-documented onychomycosis caused by C. keratinophilum in seven captive Bennett’s wallabies [ 31 ].
Diverse fungal species may occur on the skin and hairs of cats, which may be either pathogens or contaminating saprophytes. Thus, veterinary mycological diagnostics encounter dilemmas, such as contamination of superficial clinical samples by saprophytic fungi, which is most probable when the samples of hair, skin scrapings or claws are collected. Chrysosporium spp. is one of many saprophytic fungi that can contaminate the animal’s haircoat or skin and thus contribute to the contamination of clinical samples. Chrysosporium spp. has been most commonly isolated (25%) from healthy dogs and cats in Mexico [ 32 ]. This creates a challenge for veterinary laboratory diagnostics because Chrysosporium spp. shows similar characteristics to dermatophytes [ 7 ]. These fungi may have macromorphology and micromorphology similar to some Trichophyton spp., thus may be easily misidentified. Additionally, Chrysposporium spp. can grow on the DTM agar, causing pH change and redness of the medium while showing morphological characteristics corresponding to dermatophytes [ 30 ]. Furthermore, a positive hair perforation test was observed for Chrysosporium species. isolated from the environment, confirming their keratinolytic properties. Mitola et al. have described positive results of a hair perforation test for Chryspsporium georgii , Chrysosporium keratinophilum , and Chrysosporium lucknowense isolates obtained from environmental samples [ 7 ]. Likewise, Gurung et al. observed keratinolytic activity in soil isolates identified as Chrysosprium indicum and Chrysosporium fluviale [ 6 ].
A common opinion is that dermatophytes may be easily discriminated with DTM agar plate. However, literature data indicate that other fungi can also produce a positive reaction in this medium. These include Chrysosporium spp., as confirmed by Dokuzeylul et al. [ 30 ] and Jang et al. [ 33 ]. Jang et al. (2007) found that 63% of moulds isolated from dogs produced colour changes to red on DTM medium, including Chrysosporium , as well as some isolates of Aspergillus , Penicillium and others. Thus, as mentioned before, the color change of DTM agar is not sufficient to confirm the presence of dermatophytes.
The identification of our isolate with MALDI-ToF MS showed Chrysosporium keratinophilum with a high score value of 2.11. However, the sequencing of ribosomal genes indicated Chrysosporium articulatum . While performing MALDI-ToF MS analyses, the manufacturer’s Brucker database included protein spectra from only two species of this genus ( C. keratinophilum and Chrysosporium shanxiense ). Therefore, we were unable to obtain correct species identification with this method. Nevertheless, the high score value of C. keratinophilum allows us to exclude Trichophyton spp. Similar difficulties in the identification of filamentous fungi were described by Normand et al. [ 34 ] and Wilkendorf et al. [ 35 ]. The explanation for this situation is that proteomic profiles of unusual, saprophytic, filamentous fungi are currently not included in available databases, also indicating the need to expand and update these databases.
Our report describes a case of a cat with dermatological lesions initially misdiagnosed as dermatophytosis caused by Trichophyton spp. The initial identification of DTM-positive isolate as Trichophyton spp. was confirmed by colony morphology on Sabouraud agar as well as its micromorphology. Nevertheless, correct identification to the species level was obtained after sequencing of ribosomal genes. The identification using the MALDI-ToF MS technique was not possible because the available database does not include this species. Although this method allowed for the recognition of the genus Chrysosporium . Results presented in this study indicate that interpretation of the results of the mycological examination in all cases of culturing saprophytic fungi, growing from superficial samples is always challenging. Thus, careful consideration of the primary causative agent of the clinical lesions observed on the skin is mandatory. Moreover, DTM medium should be used only as a screening method, and the identification of DTM-positive isolates as dermatophytes must be confirmed by other tests.
Data availability
The dataset generated and analyzed during the current study is available in the NCBI GenBank repository, under the accession number: PP758650.
Abbreviations
Matrix assisted laser desorption ionization-time of flight mass spectrometry
Sabouraud dextrose agar (SDA); DTM - Dermatophyte test medium
Cafarchia C, Romito D, Sasanelli M, Lia R, Capelli G, Otranto D. The epidemiology of canine and feline dermatophytoses in southern Italy. Mycoses. 2004;47(11–12):508–13. https://doi.org/10.1111/j.1439-0507.2004.01055.x .
Article PubMed CAS Google Scholar
Dworecka-Kaszak B, Biegańska MJ, Dąbrowska I. Occurrence of various pathogenic and opportunistic fungi in skin diseases of domestic animals: a retrospective study. BMC Vet Res. 2020;16(1):248. https://doi.org/10.1186/s12917-020-02460-x .
Article PubMed PubMed Central CAS Google Scholar
Chupia V, Ninsuwon J, Piyarungsri K, et al. Prevalence of Microsporum canis from Pet cats in Small Animal hospitals, Chiang Mai, Thailand. Vet Sci. 2022;9(1):21. https://doi.org/10.3390/vetsci9010021 .
Article PubMed PubMed Central Google Scholar
Jarjees KI, Issa NA. First study on molecular epidemiology of dermatophytosis in cats, dogs, and their companions in the Kurdistan region of Iraq. Vet World. 2022;15(12):2971–8. https://doi.org/10.14202/vetworld.2022.2971-2978 .
Kidd SE, Weldhagen GF. Diagnosis of dermatophytes: from microscopy to direct PCR. Microbiol 2022;Aust 43, 9–13; https://doi.org/10.1071/MA22005 .
Gurung SK, Adhikari M, Kim SW, et al. Discovery of two Chrysosporium species with keratinolytic activity from Field Soil in Korea. Mycobiology. 2018;46(3):260–8. https://doi.org/10.1080/12298093.2018.1514732 .
Mitola G, Escalona F, Salas R, García E, Ledesma A. Morphological characterization of in-vitro human hair keratinolysis, produced by identified wild strains of Chrysosporium species. Mycopathologia. 2002;156(3):163–9. https://doi.org/10.1023/a:1023340826584 .
Article PubMed Google Scholar
Database MYCOBANK. https://www.mycobank.org . Accessed 02 June 2024.
Labuda R, Bernreiter A, Hochenauer D, Kubátová A, Kandemir H, Schüller C. Molecular systematics of Keratinophyton : the inclusion of species formerly referred to Chrysosporium and description of four new species. IMA Fungus. 2021;12(1):17. https://doi.org/10.1186/s43008-021-00070-2 . PMID: 34233753; PMCID: PMC8265132.
Kandemir H, Dukik K, de Melo Teixeira M, et al. Phylogenetic and ecological reevaluation of the order Onygenales . Fungal Divers. 2022;115:1–72. https://doi.org/10.1007/s13225-022-00506-z .
Article CAS Google Scholar
Kidd S, Halliday C, Alexiou H, Ellis D. Description of medical fungi, 3rd edition. 2016, https://www.adelaide.edu.au/mycology/ua/media/1596/fungus3-book.pdf . Accessed 02 June 2024.
Suankratay C, Dhissayakamol O, Uaprasert N, Chindamporn A. Invasive pulmonary infection caused by Chrysosporium articulatum : the first case report. Mycoses. 2015;58(1):1–3. https://doi.org/10.1111/myc.12270 .
Sigler L, Hambleton S, Paré JA. Molecular characterization of reptile pathogens currently known as members of the Chrysosporium anamorph of Nannizziopsis Vriesii complex and relationship with some human-associated isolates. J Clin Microbiol. 2013;51(10):3338–57. https://doi.org/10.1128/JCM.01465-13 .
Lott MJ, Moore RL, Milic NL, Power M, Shilton CM, Isberg SR. Dermatological conditions of farmed crocodilians: a review of pathogenic agents and their proposed impact on skin quality. Vet Microbiol. 2018;225:89–100. https://doi.org/10.1016/j.vetmic.2018.09.022 .
Saidi SA, Bhatt S, Richard JL, Sikdar A, Ghosh GR. Chrysosporium Tropicum as a probable cause of mycosis of poultry in India. Mycopathologia. 1994;125(3):143–7. https://doi.org/10.1007/BF01146518 .
Yamaguchi S, Sano A, Hiruma M, et al. Isolation of dermatophytes and related species from domestic fowl (Gallus gallus Domesticus). Mycopathologia. 2014;178(1–2):135–43. https://doi.org/10.1007/s11046-014-9758-0 .
Boehm TMSA, Mueller RS. Dermatophytosis in dogs and cats - an update. Dermatophytose Bei Hund Und Katze – Ein Update. Tierarztl Prax Ausg K Kleintiere Heimtiere. 2019;47(4):257–68. https://doi.org/10.1055/a-0969-1446 .
Pin D. Non-dermatophyte dermatoses mimicking dermatophytoses in animals. Mycopathologia. 2017;182(1–2):113–26. https://doi.org/10.1007/s11046-016-0090-8 .
Frymus T, Gruffydd-Jones T, Pennisi MG, et al. Dermatophytosis in cats: ABCD guidelines on prevention and management. J Feline Med Surg. 2013;15(7):598–604. https://doi.org/10.1177/1098612X13489222 .
Hernandez-Bures A, Pieper JB, Bidot WA, O’Dell M, Sander WE, Maddox CW. Survey of dermatophytes in stray dogs and cats with and without skin lesions in Puerto Rico and confirmed with MALDI-TOF MS. PLoS ONE. 2021;16(9):e0257514. https://doi.org/10.1371/journal.pone.0257514 .
Brillowska-Dabrowska A, Saunte DM, Arendrup MC. Five-hour diagnosis of dermatophyte nail infections with specific detection of Trichophyton Rubrum . J Clin Microbiol. 2007;45(4):1200–4. https://doi.org/10.1128/JCM.02072-06 .
White TJ, Bruns T, Lee S, Taylor JW. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. New York: Academic Press Inc.; 1990. pp. 315–22.
Google Scholar
Rochette F, Engelen M, Vanden Bossche H. Antifungal agents of use in animal health–practical applications. J Vet Pharmacol Ther. 2003;26(1):31–53. https://doi.org/10.1046/j.1365-2885.2003.00457.x .
Moriello K. 2019. Dermatophytosis in cats and dogs: a practical guide to diagnosis and treatment. In Practice. 2019;41:138–147; https://doi.org/10.1136/inp.l1539 .
Anstead GM, Sutton DA, Graybill JR. Adiaspiromycosis causing respiratory failure and a review of human infections due to Emmonsia and Chrysosporium spp. J Clin Microbiol. 2012;50(4):1346–54. https://doi.org/10.1128/JCM.00226-11 .
Elad D. Disseminated canine mold infections. Vet J. 2019;243:82–90. https://doi.org/10.1016/j.tvjl.2018.11.016 .
Cook E, Meler E, Garrett K, et al. Disseminated Chrysosporium infection in a German shepherd dog. Med Mycol Case Rep. 2016;10:29–33. https://doi.org/10.1016/j.mmcr.2016.01.002 .
Watt PR, Robins GM, Galloway AM, O’Boyle DA. Disseminated opportunistic fungal disease in dogs: 10 cases (1982–1990). J Am Vet Med Assoc. 1995;207(1):67–70.
Scott EM, Carter RT. Canine keratomycosis in 11 dogs: a case series (2000–2011). J Am Anim Hosp Assoc. 2014;50(2):112–8. https://doi.org/10.5326/JAAHA-MS-6012 .
Dokuzeylul B, Basaran Kahraman B, Sigirci BD, Gulluoglu E, Metiner K, Or ME. Dermatophytosis caused by a Chrysosporium species in two cats in Turkey: a case report. Vet Med-Czech. 2013;5:633–46. https://doi.org/10.17221/7187-VETMED .
Article Google Scholar
Pin D, Vidémont E, Derian-Autier D, Guillot J, Plouzeau E. First description of onychomycosis caused by Chrysosporium Keratinophilum in captive Bennett’s wallabies (Macropus rufogriseus rufogriseus). J Zoo Wildl Med. 2011;42(1):156–9. https://doi.org/10.1638/2010-0129.1 .
Guzman-Chavez RE, Segundo-Zaragoza C, Cervantes-Olivares RA, Tapia-Perez G. Presence of keratinophilic fungi with special reference to dermatophytes on the haircoat of dogs and cats in México and Nezahualcoyotl cities. Rev Latinoam Microbiol. 2000;42(1):41–4.
PubMed CAS Google Scholar
Jang KS, Yun YH, Yoo HD, Kim SH. Molds isolated from pet dogs. Mycobiology. 2007;35(2):100–2. https://doi.org/10.4489/MYCO.2007.35.2.100 .
Normand AC, Cassagne C, Gautier M, et al. Decision criteria for MALDI-TOF MS-based identification of filamentous fungi using commercial and in-house reference databases. BMC Microbiol. 2017;17(1):25. https://doi.org/10.1186/s12866-017-0937-2 .
Wilkendorf LS, Bowles E, Buil JB, et al. Update on Matrix-assisted laser desorption ionization-time of Flight Mass Spectrometry Identification of Filamentous Fungi. J Clin Microbiol. 2020;58(12):e01263–20. https://doi.org/10.1128/JCM.01263-20 .
Download references
Acknowledgements
The authors would like to thank Beata Kowalkowska for her excellent technical assistance.
Studies were partially financed by the Science Development Foundation – Warsaw University of Life Sciences.
Author information
Authors and affiliations.
Department of Preclinical Sciences, Institute of Veterinary Medicine, Warsaw University of Life Sciences-SGGW, Ciszewskiego Str. 8, Warsaw, 02-786, Poland
Magdalena Kizerwetter-Świda, Iwona Bąk, Małgorzata Justyna Biegańska & Dorota Chrobak-Chmiel
Department of Small Animal Diseases and Clinic, Institute of Veterinary Medicine, Warsaw University of Life Sciences-SGGW, Nowoursynowska 159c, Warsaw, 02-776, Poland
Kourou Dembele
You can also search for this author in PubMed Google Scholar
Contributions
DCC obtained all clinical samples, prepared all photographs, and provided contact with the cat’s owner and a veterinarian. DCC and MKŚ performed phenotypic identification, DNA isolation, PCR and sequencing analysis. MJB and IB were involved in mycological consultation. IB provided valuable comments regarding PCR and sequencing. KD conducted the clinical examination and differential diagnosis. All authors have read, critically discussed the results, and approved the manuscript.
Corresponding author
Correspondence to Magdalena Kizerwetter-Świda .
Ethics declarations
Ethical approval.
This study did not require the approval of an ethical committee since it is a case report.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Kizerwetter-Świda, M., Bąk, I., Biegańska, M.J. et al. Chrysosporium articulatum mimicking Trichophyton spp. infection in a cat: a case presentation and literature review. BMC Vet Res 20 , 359 (2024). https://doi.org/10.1186/s12917-024-04185-7
Download citation
Received : 17 April 2024
Accepted : 08 July 2024
Published : 10 August 2024
DOI : https://doi.org/10.1186/s12917-024-04185-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Dermatophytosis
- Chrysosporium articulatum
- Trichophyton spp.
- Misidentification
BMC Veterinary Research
ISSN: 1746-6148
- General enquiries: [email protected]
ORIGINAL RESEARCH article
Analyzing three pedigrees in x-linked alport syndrome with the presentation of nephrotic syndrome.

- 1 Department of Nephrology and Traditional Chinese Medicine, Shengli Clinical Medical College of Fujian Medical University, Fujian Provincial Hospital, Fuzhou, China
- 2 Department of Digestive Endoscopy, Fujian Provincial Hospital, Fuzhou, China
- 3 Department of Nephrology, Fujian Provincial Hospital, Fuzhou, China
- 4 Department of Hematology, Fujian Provincial Hospital, Fuzhou, China
Background: Alport syndrome (AS) is a common cause of end-stage renal disease (ESRD) with various clinical symptoms and incomplete manifestation. Patients with AS and other renal disorders are often misdiagnosed. This study reported three X-linked dominant Alport syndrome (XLAS) pedigrees with nephrotic syndrome (NS) as the predominant phenotype and analyzed COL4A5 gene alterations.
Methods: Three Han Chinese XLAS pedigrees were recruited, and clinical phenotypes were obtained. The pre-certified individuals’ peripheral blood DNA was taken, and whole-genome next-generation sequencing (NGS) was performed for candidate genes and mutation screening, followed by NGS or Sanger sequencing of suspected mutant types in participating family members.
Results: Both probands A and B were diagnosed with NS through biochemical tests, and X-linked Alport syndrome-associated renal injury was diagnosed by renal biopsy. The biopsy revealed focal foamy cells in the renal interstitium, tearing and delamination changes in the glomerular basement membrane, and negative α3 and α5 chains of type IV collagen. Proband C, who was earlier diagnosed with NS, has now advanced to ESRD, along with his mother and proband A’s mother. Genetic sequencing of all three pedigrees identified three mutations, namely, c.5020C>T, c.4435_4445del, and c.1584_1587+6del in the X-linked dominant gene COL4A5 (NM_000495.5). These mutations lead to the production of shortened proteins, potentially impacting the function of COL4A5 and causing pathogenic effects.
Conclusion: The novel c.4435_4445del and c.1584_1587+6del mutations not only enrich the spectrum of mutations in the COL4A5 gene but also indicate that carriers of both mutation sites and those with mutation c.5020C>T may present NS as their primary clinical manifestation.
Introduction
Alport syndrome (AS) is a prevalent genetic kidney disease known as hereditary nephritis and oculo-auriculo-renal syndrome. It is caused by mutations in the COL4A3 , COL4A4 , and COL4A5 genes, which encode the α3, α4, and α5 chains of type IV collagen. X-linked dominant, autosomal recessive, or autosomal dominant inheritance are the modes of inheritance of AS, which causes hematuria, proteinuria, sensorineural deafness, ocular abnormalities, and kidney damage that leads to end-stage renal disease (ESRD) ( Daga et al., 2022 ). X-linked Alport syndrome (XLAS, OMIM: 301050) is caused by a pathogenic variant in the COL4A5 gene and is the most common mode of inheritance. Men have up to a 100% risk of progression to ESRD, and the age of progression and extrarenal manifestations correlate with the genotype; women have a 25% risk and an increased risk with age ( Alport Syndrome Collaborative, National Clinical Research Center of Kidney, & Rare Diseases Branch of Beijing Medical, 2023 ). A history of childhood hematuria, steadily deteriorating proteinuria, and sensorineural deafness are risk factors for the advancement of this illness ( Jais et al., 2003 ).
Patients with AS can exhibit various clinical symptoms, which may not include the typical signs during the initial stages. Consequently, when AS occurs alongside other kidney diseases, patients may be mistakenly diagnosed with IgA nephropathy, nephrotic syndrome (NS), purpura nephritis, or thin basement membrane nephropathy ( Deng, Zhou and Zhou, 2023 ). Cases of AS with NS as its primary symptom are infrequently documented, leading to the potential oversight of AS and subsequent delays in diagnosis, ultimately culminating in the progression to ESRD. This article presents three pedigrees of XLAS resulting from variations in the COL4A5 gene, with the identification of two novel mutation sites. Unexpectedly, all three probands presented with NS as their primary clinical symptom, with two of them experiencing NS as their initial symptom. We conducted genetic and phenotypic analyses of the three pedigrees, highlighting the crucial nature of renal pathological examination and genetic testing in diagnosing atypical kidney diseases.
Method and material
Research subjects.
Three Chinese pedigrees with Alport syndrome were collected in the study: 12 people from pedigree A, 13 people from pedigree B, and 14 people from pedigree C. Within the pedigrees, there were no consanguineous marriages ( Figures 1A–C ). The Fujian Provincial Hospital Ethics Committee approved the study. The pedigree members willingly signed an informed consent form before the clinical investigation. Proband A (A-III6) and proband B (B-III2) experienced a sudden onset of illness, characterized mostly by edema of the eyelids and both lower limbs, together with hematuria and foamy urine, but with normal urine output. When proband C (C-III5) was 3 years old, he began to exhibit unclear-source hematuria, and the renal pathology at that time pointed to diffuse proliferative glomerulonephritis with a few segmental scleroses. At the age of 17 years, he suddenly developed swelling of the eyelids and the lower limbs, with serum albumin of 20 g/L and a large amount of proteinuria, which was diagnosed as “nephrotic syndrome.” A second renal biopsy suggested proliferative glomerulonephritis with focal and segmental glomerulosclerosis (FSGS), and he has been on maintenance peritoneal dialysis for more than 10 years because of his ESRD. Probands A and C, whose mothers (A-II6 and C-II5) are presently receiving hemodialysis for ESRD, have a medical history of frothy urine and hematuria. Apart from a few members of pedigree A (II6 and III6), none of the respondents had hearing impairment, and none of the pedigrees had reported ocular pathology. Other members of the three pedigrees were examined for past and present disease histories, as well as for general physical examinations, alongside genetic testing ( Figure 1D ).

Figure 1 . (A) Pedigree A map, black representing carriers of the COL4A5 (NM_000495.5) c.5020C>T mutation, with red arrows representing pre-documented individuals, squares representing male population, and circles representing female population; (B) pedigree B map, black representing carriers of the COL4A5 (NM_000495.5) c.4435_4445del mutation; (C) pedigree C map, black representing COL4A5 (NM_000495.5) c.1584_1587+6del mutation carriers. (D) The documented presence of clinical symptoms in the carriers of the mutant in three pedigrees.
DNA extraction
Peripheral blood samples (5 mL) were taken from the proband and family members and collected in an anticoagulant tube using ethylenediaminetetraacetic acid (Shanghai Solarbio Bioscience & Technology Co., Ltd., Shanghai, China), and the genome was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany). DNA concentration and purity were measured using a NanoDrop 1000 Spectrophotometer (NanoDrop Technologies, United States).
Candidate gene location and mutation-screening strategies
After testing for DNA quality, whole-genome sequencing was performed, and based on bam results of comparison with genome reference sequences, SAMtools, GATK, and ANNOVAR were used to find and analyze SNVs and indels in the sequencing data and screen and annotate variants according to ACMG ( Richards et al., 2015 ) in databases such as dbSNP, 1000G, HGMD, and ESP6500. Sequence reads were compared with hg19 sequences using BWA to analyze gene information, mutation types, frequencies from databases such as 1000G and ESP6500. Mutation pathogenicity was predicted using software like PolyPhen-2, Sorting Intolerant from Tolerant, and MutationTaster. Primer Premier 5.0 was used to create amplification primers for the upstream and downstream regions of sequences with target mutation sites, enabling the amplification of target regions.
Sanger sequencing verification
PCR amplification and Sanger sequencing were used to validate the candidate mutation sites in the proband and the pedigrees. Using the GenBank (NM_000495.5) COL4A5 gene sequence, Primer Premier5.0 software designed primers for the target sequences. The primers were synthesized by Synbio Technologies Co., Ltd. (Suzhou) ( Table 1 ). PCR products were amplified and purified using the reagent from TaKaRa (Takara Biomedical Technology Co., Ltd., Beijing) and sequenced using an ABI 3730XL sequencer (Beijing Jingkreida Technology Co., Ltd., Beijing) to detect the PCR products of target fragments.

Table 1 . Primer design of the fragments with suspected responsibility point mutation of each Alport syndrome pedigree.
Clinical phenotypes
Proband A (A-III6) and proband B (B-III2) both exhibited hypoproteinemia and massive proteinuria ( Table 2 ), and they were diagnosed with nephrotic syndrome. Proband C (C-III5) and his mother (C-II5) and proband A’s mother (A-II6) had glomerular filtration rates below 10 mL/min, leading to the diagnosis of ESRD. Additionally, they experienced renal anemia and serum electrolyte disorders ( Table 2 ). Furthermore, proband A’s grandmother (A-Ⅰ2) and uncle (A-II1) succumbed to renal illness. Renal-related biochemical markers did not exhibit any abnormalities in the remaining members of pedigrees A and C (A-III1, A-III5, C-II3, C-II7, and C-III2). Probands A and B underwent ultrasound-guided percutaneous kidney puncture biopsies with informed consent. Immunofluorescence analysis of renal pathology in proband A showed IgM positivity, weak positivity for IgA and C3, negativity for IgG and C1q, and positivity for type IV collagen staining α1, but negativity for α3 and α5( Figures 2A–D ). Electron microscopy revealed that the glomerular basement membrane exhibited varying thickness, ranging from approximately 150 to 390 nm. Additionally, the membrane displayed tears, layered alterations, a diffuse fusion of podocyte foot processes, mild hyperplasia of the tethered cells, and stroma. Notably, there was an absence of electron-dense material in the subepithelial region, interior of the basement membrane, or subendothelial area. The renal tubular epithelial cells display vacuolar degeneration, together with a minor presence of inflammatory cells in the renal interstitium ( Figures 2E–G ). A light microscopic stain showed a small amount of photophilic protein deposition in the mesangial area, inadequate capillary collateral dilatation, uneven basement membrane staining, and modest hyperplasia of certain glomerular mesangial cells and stroma. Renal tubular epithelial cells, vacuolar degeneration, focal inflammatory cell infiltration restricted by the renal interstitium, no discernible fibrosis, focal foam-like cells visible in the renal interstitium, and no visible lesions in the small arteriolar wall ( Figures 2H–K ). Immunofluorescence analysis of proband B suggested weak positivity for IgM and C3, negativity for IgG, IgA, C1q, Fib, and ALB, and positivity for α1; α3, α4, and α5 expressions were absent ( Figures 2L–O ). Electron microscopy discovered that the thickness of the glomerular basement membrane varied between 100 and 300 nm. The dense layer of the membrane had a stratified structure, with incomplete inner and outer edges resembling a flower basket. There was an increase in the number of mesangial cells and stroma in the area where the glomerulus is attached. The podocyte foot process pedicles showed widespread fusion. Some glomerular attachment sites contained a small amount of electron-dense material. Additionally, there was metaplasia of the vacuoles in the tubular epithelial cells. No distinct abnormalities were observed in the renal interstitium ( Figures 2P–R ). Extracapsular fibrosis and thickening of the glomerular capsule wall were visible under light microscopy. The features that are visible in the renal tubules include dilated lumens in most of them, brush border loss, diffuse clusters of foamy cell infiltration, renal tubular epithelial cell vacuolation, granular degeneration, multiple tubular foamy degenerations, visible protein tubular patterns, renal interstitial edema, and mild fibrosis of the interstitium ( Figures 2S–V ). The renal biopsies of probands A and B revealed the presence of foam-like cells in the renal interstitium and ripping and layering abnormalities in the glomerular basement membrane. Additionally, the staining for type IV collagen was negative for both α3 and α5; therefore, XLAS renal injury was considered.

Table 2 . Clinical data of the members in every family associated with Alport syndrome.

Figure 2 . Renal pathological findings of proband A: (A) weakly positive immunofluorescence with IgA; (B) positive type IV collagen α1 staining; (C) negative type IV collagen α3 staining; (D) negative type IV collagen α5 staining; (E–G) electron microscopy revealed glomerular basement membranes of varying thicknesses and thinnesses, basement membrane tearing, layered alterations, a diffuse fusion of podocyte foot processes, and mild proliferation of the glomerular mesangial cells and stroma; (H,I) light microscopy using H&E and PAS staining revealed mild hyperplasia of some glomerular mesangial cells and stroma, vacuolar degeneration of renal tubular epithelial cells, small foci of inflammatory cell infiltration in the renal interstitial, and foci of foamy cells (200×; 400×); (J) PASM staining did not reveal peg-like structures, thylakoid insertion, or double track formation (400×); (K) Masson staining did not reveal a distinct lesion (400×). The renal pathological findings of proband B: (L) positive immunofluorescence α1; (M) negative type IV collagen staining α3; (N) negative type IV collagen staining α4; and (O) negative type IV collagen staining α5; (P–R) electron imaging revealed the stratification of the dense layer of the glomerular basement membrane with incomplete inner and outer rims in the form of a flower basket and proliferation of the glomerular tethered area of the tethered cells and stroma; (S,T) the glomerular capsule wall thickened, and extracapsular fibrosis, vacuolar and granular degeneration of renal tubular epithelial cells, multiple tubular foamy degeneration, visible protein tubular pattern, interstitial edema, focal inflammatory cell infiltration, diffuse clusters of foamy cell infiltration, and mild fibrosis of the interstitium were all revealed by light microscopic H&E and PAS staining (200× and 400×); (U) PASM staining failed to reveal peg-like structures without tethered membrane insertion and double-track formation (400×); (V) Masson’s staining did not reveal any obvious lesions (400×).
Screening for mutations in the Alport syndrome gene
Each proband had one mutant site and was hemizygous with X-linked dominant inheritance according to whole-genome sequencing. Proband A has the mutation site c.5020C>T in exon53 of COL4A5 (NM_000495.5), which turns the arginine at position 1,674 into a termination codon (p.Arg1674Ter) and truncates the protein. Proband B has a deletion mutation in exon47 of COL4A5 (NM_000495.5) that produces p.Thr1480Leufs* 2, caused by the deletion of 11 bases from positions 4,435 to 4,445. Proband C has a deletion mutation in exon23 of COL4A5 (NM_000495.5) that deletes bases 1,584–1,587 and six intronic bases (c.1584_1587+6del), causing the termination codon to occur early and creating a shortened protein. Whole-genome sequencing was performed on all members of pedigree A upon special request. The results showed that A-II6, A-III1, A-III5, and A-III6 carry the mutation c.5020C>T. Given that this mutation (p. Arg1674Ter) has been reported and considered pathogenic ( Zhang et al., 2018 ), we hypothesize that the two members of the pedigree who passed away from renal disease (A-Ⅰ2 and A-Ⅱ1) may also carry this mutation. The remaining pedigree B and C members were Sanger-sequenced. In pedigree B, only the proband has the c.4435_4445del (p.Thr1480Leufs* 2) mutation, while the rest of the members are wild type ( Figures 3A, B ). In pedigree C, Ⅰ2, Ⅱ3, Ⅱ5, Ⅱ7, Ⅲ2, and Ⅲ5 carry the c.1584_1587+6del mutation, while in others, no mutants were found ( Figures 3C–E ). As per the ACMG ( Richards et al., 2015 ), the mutation c.5020C>T is classified as pathogenic based on the criteria PVS1_Strong + PS4_Supporting + PM2_Supporting. Similarly, the mutation c.4435_4445del is also considered pathogenic according to the criteria PVS1_Strong + PS2 + PM2_Supporting. On the other hand, the mutation c.1584_1587+6del is classified as possibly pathogenic based on the criteria PVS1 + PM2_Supporting.

Figure 3 . (A) Sanger sequencing map of pedigree B, COL4A5 (NM_000495.5) c. 4435_4445del hemizygous mutant; (B) corresponding wildtype of pedigree B; (C) Sanger sequencing map of pedigree C, COL4A5 (NM_000495.5) c.1584_1587+6del hemizygous mutant; (D) corresponding heterozygous mutant; (E) corresponding wild type.
Prediction and analysis of bioinformatics
Our investigation revealed that all three mutants generate shortened proteins ( Figures 4A–C ) due to the premature occurrence of the stop codon, and all of them are classified as pathogenic. To verify this hypothesis, we employed SWISS-MODEL ( https://swissmodel.expasy.org/repository/uniprot/P29400 ) to forecast the 3D configurations of the proteins belonging to the wild-type (WT) and COL4A5 mutations. Subsequently, we displayed the three-dimensional structure image of the COL4A5 WT using PyMOL ( Figure 4D ). Despite losing fewer sequences (black depicts the mutant’s missing section) ( Figure 4E ), the protein’s three-dimensional structures have changed ( Figure 4F ). c.4435_4445del and c.1584_1587+6del mutants lack additional sequences ( Figures 4G, I ). Mutated proteins had very different three-bit structures ( Figures 4H, J ).

Figure 4 . (A–C) Amino acid sequence changes following the three mutations c.5020C>T, c.4435_4445del, and c.1584_1587+6del of COL4A5 (NM_000495.5), all of which lead to an early termination codon and end of translation of the protein COL4A5. The tertiary structure of COL4A5 wildtype and mutants were predicted using SWISS-MODEL ( https://swissmodel.expasy.org/repository/uniprot/P29400 ) and shown in PyMOL. (D) Tertiary structure of COL4A5 wildtype protein; (E) black part represents the protein structure of mutant c.5020C>T deletion; (F) tertiary structure of mutant c.5020C>T protein; (G) black represents the protein structure of mutant c.4435_4445del deletion; (H) tertiary structure of mutant c.4435_4445del protein; (I) black part represents the missing protein structure of mutation c.1584_1587+6del; (J) protein 3D structure of mutant c.1584_1587+6del.
As estimated, 1/10,000–1/5,000 people have AS, which accounts for 0.5% of newly diagnosed adult cases of ESRD ( Mallett et al., 2014 ). Nonetheless, it was shown that COL4A5 variants with potential for disease were often found in renal failure queues and normal reference data, suggesting that the occurrence rate of XLAS was around 1/5,000 ( Groopman et al., 2019 ; Connaughton and Hildebrandt, 2020 ). This supports AS being the second leading cause of hereditary renal failure after autosomal dominant polycystic kidney disease ( Savige et al., 2021 ). Nephrotic syndrome is a rare but dangerous kidney illness that affects people worldwide, including adults and children. Edema, proteinuria, hypoalbuminemia, and hyperlipidemia are common symptoms (C. S. Wang and Greenbaum, 2019 ). Incidence rates are 3 per 100,000 adults and 2–7 per 100,000 children annually. Although it occurs rarely, it is responsible for 20% of pediatric ESRD cases and roughly 12% of all ESRD cases ( Verma and Patil, 2024 ). Numerous conditions can lead to NS, including primary glomerulonephritis, infections, tumors, and drug-induced illnesses. In our pedigree study, all three probands met the diagnostic criteria for the nephrotic syndrome: albumin levels less than 30 g/L, 24-h urine protein levels more than 3.5 g/L, edema, and/or high lipid levels ( Kodner, 2016 ). We did not delay performing renal puncture for the two patients with AS whose first manifestation was nephrotic syndrome; however, proband C was not diagnosed with AS in a timely manner, probably because of limited medical technology or a lack of awareness of AS at that time. He was also not found to carry the COL4A5 mutant until he progressed to ESRD by genetic testing.
Nephrotic syndrome primarily arises from the impairment or malfunction of glomerular components, such as the glomerular basement membrane, endothelial surface, or epithelial cells (podocytes) ( Politano, Colbert, and Hamiduzzaman, 2020 ). This results in the excretion of protein in the urine and encompasses three primary types of pathology: microscopic lesion disease, FSGS, and idiopathic membranous nephropathy. Among these, FSGS is the primary cause of ESRD in nephrotic syndrome ( Ozeki et al., 2021 ). Furthermore, AS leads to the development of either segmental or widespread mesangial cell hyperplasia, accompanied by an increase in mesangial stroma as the disease advances, and some cases may present glomerulopathy with FSGS ( Alport Syndrome Collaborative et al., 2023 ). Proband C’s second renal pathology was diagnosed as mesangial proliferative glomerulonephritis with FSGS, which aligns with AS-related renal pathology. The patient experienced prolonged hematuria starting at the age of 3, which is more likely to be attributed to an inherited nephrotic disease-related nephrotic syndrome in a pediatric patient with a family history of the disease. In the remaining two probands, electron microscopy revealed tearing and layering alterations in the basement membrane of the kidneys. Additionally, immunofluorescence analysis showed a lack of α3 and α5 collagen staining, which aligns with the characteristic renal damage observed in individuals with AS. Therefore, both patients can be classified as having AS-related nephrotic syndrome. What sets these previous patients apart is that they not only exhibit the classic symptoms of nephrotic syndrome, but they also have hematuria. Hematuria can be a significant clinical sign of nephrotic syndrome when combined with AS. A case report from China details the presentation of an 11-month-old infant with AS who primarily exhibited nephrotic syndrome (D. Wang et al., 2021 ). Additionally, the child experienced extensive hematuria, but her renal function remained unaffected. Another Chinese child diagnosed with AS exhibited nephrotic syndrome as the major symptom, along with the presence of microscopic hematuria ( Deng et al., 2023 ). Therefore, we should not assume that hematuria is solely caused by nephrotic syndrome since it may serve as a crucial indicator in our investigation of the underlying cause of nephrotic syndrome.
Undoubtedly, type IV collagen α-chain component anomalies in probands A and B necessitate genetic testing. Furthermore, patients should have undergone COL4A5 genetic testing if they have persistently abnormal hematuria for more than 6 months or persistent proteinuria greater than 0.5 g per day in addition to renal biopsy confirmation of FSGS ( Savige et al., 2022 ), so proband C should have undergone genetic testing much earlier. Through genetic testing, it was determined that all three probands were XLAS patients. Among male XLAS patients, there was a significant correlation between their genetic makeup and the severity of their kidney symptoms. Patients with truncating variants, such as nonsense variants or small insertions/deletions that cause premature termination of codons, exhibited more severe symptoms compared to patients with missense variants or small in-frame variants ( Yamamura et al., 2020 ). As of the current date (2024-03-14), the ClinVar ( https://www.ncbi.nlm.nih.gov/clinvar ) database includes 192 frameshift mutations and 113 nonsense mutations, all of which are classified as pathogenic or likely pathogenic. Out of the three mutations we found, mutation c.5020C>T is harmful and associated with bilateral symmetrical sensorineural deafness ( Zhang et al., 2018 ). Coincidentally, both proband A and his mother, who also has the mutation c.5020C>T, have a simultaneous combination of hearing impairment. Patients with missense mutations have a 60% risk of hearing loss by the age of 30, while patients with other types of mutations have a 90% risk ( Jais et al., 2000 ). Most patients with hearing loss experience mild to moderate hearing loss, which worsens over time. The other two families, although currently unaffected, will need ongoing hearing tests. The mutation sites c.4435_4445del and c.1584_1587+6del have not been previously documented. The ACMG recommendations classify c.4435_4445del as pathogenic and c.1584_1587+6del as probabilistically pathogenic. We conducted a three-dimensional protein structure analysis and discovered that both mutations result in the production of shortened proteins. The region that is absent encompasses not just the specific mutation site c.5020C>T but also extends beyond it. Therefore, we hypothesize that both mutations may have an impact on the protein’s function and are likely to be pathogenic.
Currently, there is no definitive remedy for AS. The treatment primarily aims to decrease proteinuria and slow down the advancement of kidney disease. Although treatment can postpone the start of renal impairment, most AS patients will eventually require dialysis or renal transplantation. For men diagnosed with XLAS, it is advisable to start renin–angiotensin–aldosterone system (RAAS) blocker medication ( Kashtan and Gross, 2021 ). Probands A and B were instructed to take oral valsartan and dapagliflozin to decrease the excretion of protein in the kidneys. Furthermore, both patients had the additional condition of nephrotic syndrome. After taking glucocorticoids, there was a considerable improvement in edema for both patients, and adjuvant medications such as compound α-ketoacid, calcium, gastric preservation medication, and atorvastatin were utilized. Moreover, the two probands’ continued to experience hematuria while showing improvement in proteinuria during the follow-up. The fact that glucocorticoid therapy usually has no effect on nephrotic syndrome associated with Alport syndrome (D. Wang et al., 2021 ) leads us to consider that the two patients may have combined minimal change nephrotic syndrome. Proband C and the mother of both probands had reached ESRD and needed renal replacement treatment.
New mutations c.4435_4445del and c.1584_1587+6del enriched the COL4A5 gene mutation spectrum, and carriers of these two mutation sites and c.5020C>T may present nephrotic syndrome as the predominant clinical symptom. Renal pathological examination and genetic testing are crucial to diagnosing AS when NS is the major indication. This is because the standard renal signs are lost, making it easy to detect or misdiagnose.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/ , SCV004698203 https://www.ncbi.nlm.nih.gov/ , SCV004698204 https://www.ncbi.nlm.nih.gov/ , SCV004698332.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
J-HZ: writing–original draft. JL: writing–original draft. D-DR: writing–original draft. QC: writing–original draft. JY: writing–original draft. MW: writing–original draft. H-PY: writing–original draft. L-SL: writing–review and editing. X-LZ: writing–review and editing. J-WL: writing–review and editing. LZ: writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Fujian Province Medical Innovation Foundation (2022CXA001,2022CXB002, 2021CXB001), the Fujian Province Natural Science Fund Project (2022J01409, 2021J02053, 2023J011159, 2022J01996), the grants from Joint Funds for the innovation of science and Technology in Fujian province (2023Y9284), the Special Research Foundation of Fujian Provincial Department of Finance (No. 2023-830, 2022-840), and National famous and old Chinese medicine experts (Xuemei Zhang, Xiaohua Yan, Shaoguang Lv) inheritance studio construction project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alport Syndrome Collaborative, Group, National Clinical Research Center of Kidney, Diseases, & Rare Diseases Branch of Beijing Medical, AssociationNational Clinical Research Center of Kidney DiseasesRare Diseases Branch of Beijing Medical Association (2023). Zhonghua Yi Xue Za Zhi 103 (20), 1507–1525. [expert consensus on the diagnosis and treatment of alport syndrome (version 2023)]. doi:10.3760/cma.j.cn112137-20230203-00161
PubMed Abstract | CrossRef Full Text
Connaughton, D. M., and Hildebrandt, F. (2020). Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol. Dial. Transpl. 35 (3), 390–397. doi:10.1093/ndt/gfz028
PubMed Abstract | CrossRef Full Text | Google Scholar
Daga, S., Ding, J., Deltas, C., Savige, J., Lipska-Zietkiewicz, B. S., Hoefele, J., et al. (2022). The 2019 and 2021 international workshops on alport syndrome. Eur. J. Hum. Genet. 30 (5), 507–516. doi:10.1038/s41431-022-01075-0
Deng, Z., Zhou, Q., and Zhou, T. G. (2023). A case report and literature study on alport syndrome featuring nephrotic syndrome as its primary manifestation. Transpl. Immunol. 81, 101941. doi:10.1016/j.trim.2023.101941
Groopman, E. E., Marasa, M., Cameron-Christie, S., Petrovski, S., Aggarwal, V. S., Milo-Rasouly, H., et al. (2019). Diagnostic utility of exome sequencing for kidney disease. N. Engl. J. Med. 380 (2), 142–151. doi:10.1056/NEJMoa1806891
Jais, J. P., Knebelmann, B., Giatras, I., De Marchi, M., Rizzoni, G., Renieri, A., et al. (2003). X-linked alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "european community alport syndrome concerted action" study. J. Am. Soc. Nephrol. 14 (10), 2603–2610. doi:10.1097/01.asn.0000090034.71205.74
Jais, J. P., Knebelmann, B., Giatras, I., Marchi, M., Rizzoni, G., Renieri, A., et al. (2000). X-linked alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 11 (4), 649–657. doi:10.1681/ASN.V114649
Kashtan, C. E., and Gross, O. (2021). Clinical practice recommendations for the diagnosis and management of alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr. Nephrol. 36 (3), 711–719. doi:10.1007/s00467-020-04819-6
Kodner, C. (2016). Diagnosis and management of nephrotic syndrome in adults. Am. Fam. Physician 93 (6), 479–485.
PubMed Abstract | Google Scholar
Mallett, A., Tang, W., Clayton, P. A., Stevenson, S., McDonald, S. P., Hawley, C. M., et al. (2014). End-stage kidney disease due to alport syndrome: outcomes in 296 consecutive Australia and New Zealand dialysis and transplant registry cases. Nephrol. Dial. Transpl. 29 (12), 2277–2286. doi:10.1093/ndt/gfu254
Ozeki, T., Nagata, M., Katsuno, T., Inagaki, K., Goto, K., Kato, S., et al. (2021). Nephrotic syndrome with focal segmental glomerular lesions unclassified by columbia classification; pathology and clinical implication. PLoS One 16 (1), e0244677. doi:10.1371/journal.pone.0244677
Politano, S. A., Colbert, G. B., and Hamiduzzaman, N. (2020). Nephrotic syndrome. Prim. Care 47 (4), 597–613. doi:10.1016/j.pop.2020.08.002
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Savige, J., Lipska-Zietkiewicz, B. S., Watson, E., Hertz, J. M., Deltas, C., Mari, F., et al. (2022). Guidelines for genetic testing and management of alport syndrome. Clin. J. Am. Soc. Nephrol. 17 (1), 143–154. doi:10.2215/CJN.04230321
Savige, J., Storey, H., Watson, E., Hertz, J. M., Deltas, C., Renieri, A., et al. (2021). Consensus statement on standards and guidelines for the molecular diagnostics of alport syndrome: refining the acmg criteria. Eur. J. Hum. Genet. 29 (8), 1186–1197. doi:10.1038/s41431-021-00858-1
Verma, P. R., and Patil, P. (2024). Nephrotic syndrome: a review. Cureus 16 (2), e53923. doi:10.7759/cureus.53923
Wang, C. S., and Greenbaum, L. A. (2019). Nephrotic syndrome. Pediatr. Clin. North Am. 66 (1), 73–85. doi:10.1016/j.pcl.2018.08.006
Wang, D., Shan, C., Jing, X., Zhang, Q., Chang, H., and Lin, Y. (2021). Clinical features and familial mutations in an autosomal-inherited alport syndrome patient with the presentation of nephrotic syndrome. Front. Pediatr. 9, 678633. doi:10.3389/fped.2021.678633
Yamamura, T., Horinouchi, T., Nagano, C., Omori, T., Sakakibara, N., Aoto, Y., et al. (2020). Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male x-linked alport syndrome. Kidney Int. 98 (6), 1605–1614. doi:10.1016/j.kint.2020.06.038
Zhang, X., Zhang, Y., Zhang, Y., Gu, H., Chen, Z., Ren, L., et al. (2018). X-linked alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J. Rare Dis. 13 (1), 229. doi:10.1186/s13023-018-0974-4
Keywords: Alport syndrome, COL4A5 gene, nephrotic syndrome, end-stage renal disease, pedigree analysis
Citation: Zhang J-H, Liu J, Ruan D-D, Chen Q, Yang J, Wu M, Yu H-P, Liao L-S, Zheng X-L, Luo J-W and Zhang L (2024) Analyzing three pedigrees in X-linked Alport syndrome with the presentation of nephrotic syndrome. Front. Genet. 15:1419154. doi: 10.3389/fgene.2024.1419154
Received: 17 April 2024; Accepted: 26 July 2024; Published: 09 August 2024.
Reviewed by:
Copyright © 2024 Zhang, Liu, Ruan, Chen, Yang, Wu, Yu, Liao, Zheng, Luo and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Ling Zheng, [email protected] ; Jie-Wei Luo, [email protected] ; Li Zhang, [email protected]
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
COMMENTS
The term presentation describes the leading part of the fetus or the anatomical structure closest to the maternal pelvic inlet during labor. The presentation can roughly be divided into the following classifications: cephalic, breech, shoulder, and compound. Cephalic presentation is the most common and can be further subclassified as vertex, sinciput, brow, face, and chin. The most common ...
The vast majority of fetuses at term are in cephalic presentation. Approximately 5 percent of these fetuses are in a cephalic malpresentation, such as occiput posterior or transverse, face ( figure 1A-B ), or brow ( figure 2) [ 1 ]. Diagnosis and management of face and brow presentations will be reviewed here.
Explore delivery, face presentation, and brow presentation in childbirth. Learn about the definitions, causes, complications, and management approaches for these unique fetal positions to ensure safe and successful deliveries.
7.10.2 Management. Foetus alive. Foetus dead. Brow presentation constitutes an absolute foeto-pelvic disproportion, and vaginal delivery is impossible (except with preterm birth or extremely low birth weight). This is an obstetric emergency, because labour is obstructed and there is a risk of uterine rupture and foetal distress.
Nonvertex presentations (including breech, transverse lie, face, brow, and compound presentations) occur in less than 4% of fetuses at term. Malpresentation of the vertex presentation occurs if there is deflexion or extension of the fetal head leading to brow or face presentation, respectively.
Management considerations for face, brow, and compounds presentations are unique with compound presentations having higher rates of vaginal delivery and lower complications as compared to either brow or face presentations. For brow presentations, approximately 30-40% of brow presentations will convert to a face presentation, and about 20% will ...
Our expert explains exactly what brow presentation is, the chances of it happening during your labour and how it could affect the delivery of your baby.
The causes of a brow presentation are the same as those for face presentation and may also include cephalo-pelvic disproportion and obstruction with neglected labour where the fetal head gradually becomes more and more deflexed. Alternatively, the brow presentation may itself be the cause of the labour dystocia.
The outlook of the persistent brow presentation for vaginal delivery is poor. Approximately two thirds of brow presentations will convert to vertex or face. 2 Fortunately, this is a rare presentation, with an incidence of only 0.05%. 5 If the presentation persists as a brow, a cesarean section should be performed.
Brow presentation is considered the rarest of all malpresentation with a prevalence of 1 in 500 to 1 in 4000 deliveries. Both face and brow presentations occur due to extension of the fetal neck instead of flexion; therefore, conditions that would lead to hyperextension or prevent flexion of the fetal neck can all contribute to face or brow ...
Brow presentation is the rarest of all malpresentations. Anencephaly, neck masses in fetus, polyhydramnios, multiple loops of cord around neck are the fetal factors leading to brow presentation. Contracted pelvis, preterm labour, platypelloid pelvis are some of the...
In a brow presentation, the partially extended head presents with the occipitomental diameter of 13.5 cm in the average term fetus. Brow presentations are associated with pelvic contraction, small or large fetuses, and nuchal masses. Two thirds spontaneously convert to either a face or an occipital presentation.
There are several complications associated with a brow presentation if vaginal delivery is attempted without proper measures. Increased chances of spinal cord injury are associated with brow presentation. Fetal distress. Abnormal shape of the baby's head after delivery. Prolonged labor.
Brow presentation is usually only diagnosed once labour is well established. The anterior fontanelle and super orbital ridges are palpable on vaginal examination.
In cephalic presentations, the most common malpresentation leading to intervention is a persistent occipitoposterior position ( 1,2 ). There is considerable variation in the reported frequency of some rarer cephalic malpresentations in the literature, such as brow (1:800-10 000) ( 3-5) or face presentation (1:500-2 000) ( 6,7 ).
Brow presentation is rare but can cause serious complications in pregnant women. Learn all about its causes, diagnosis, complications & more
Face presentation is a rare obstetric event and most practitioners will go through their carriers without ever meeting one. Face presentation can be delivered vaginally only if the foetus is in the mentum anterior position. More than half of the cases ...
The brow presentation may cause a redness but only occasionally will cause a bruise. Mobility of the pelvis and the freedom of maternal movements often help bring the face-first baby down through the pelvis with good strong, uterine surges.
Brow presentation 257. stitutions), which is not statistically signifi cant as an etiologic cause for the mal presentation. Seventeen instances of cephalopelvic dis proportion werc detected by clinical and/ or x-ray evaluation. This represented a 10.9 per cent incidence as compared with 2.7 per cent expected.
Introduction In previous study sessions of this module, you have been introduced to the definitions, signs, symptoms and stages of normal labour, and about the 'normal' vertex presentation of the fetus during delivery. In this study session, you will learn about the most common abnormal presentations (breech, shoulder, face or brow), their diagnostic criteria and the required actions you ...
The incidence of breech presentation is 20-25% of fetuses at <28 weeks, but only 7-16% at 32 weeks, and only 3-4% at term. 2, 3. Face and brow presentation are uncommon. Their prevalence compared with other types of malpresentations are shown below. 4. Occiput posterior - 1/19 deliveries;
8.5.1 Possible causes of brow presentation You have seen all of these factors before, as causes of other malpresentations:
Face and brow presentations are types of malpositions where the baby is positioned abnormally. In a face presentation, the baby's face is the first part to appear, while in a brow presentation, the area between the baby's forehead and the top of the skull is the leading part.
Tension headaches generally cause pain across the forehead or on both sides and the back of the head, according to the Mayo Clinic. They tend to come on slowly, and are usually mild to moderate.
We need to be bold, we need to be innovative, and we need to take advantage of this unique moment to create a transformational project not only for our fans, but for Cleveland, the Northeast Ohio ...
Here's what skin experts want you to know about cystic acne. What causes cystic acne? Cystic acne typically arises for people in their teens and 20s, but it can last into adulthood as well.
Introduction Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome of excessive immune activation that can be life-threatening. 1,2 Most commonly, HLH impacts infants, but is also observed in children and adults. Diagnosis of HLH can be challenging, as there is a large variation in presentation and severity.
The rain that drenched Friday's opening ceremony may have moved out of Paris, but its effects are still being felt in the River Seine, with water quality concerns throwing the triathlon ...
The clinical presentation includes respiratory allergic reactions, pulmonary invasive infections and skin infections. There is only one documented case of Chrysosporium articulatum invasive pulmonary infection in human, 16-year-old man diagnosed with lymphoblastic leukemia .
Background: Alport syndrome (AS) is a common cause of end-stage renal disease (ESRD) with various clinical symptoms and incomplete manifestation. Patients with AS and other renal disorders are often misdiagnosed. This study reported three X-linked dominant Alport syndrome (XLAS) pedigrees with ...